


Significant outcomes
∙ Adjunctive brexpiprazole did not differentiate from antidepressant therapy (ADT)+placebo on the primary endpoint of full remission.
∙ Brexpiprazole was well tolerated in patients with major depressive disorder (MDD) when administered as adjunctive therapy for 24 weeks, with no unexpected side effects.
Limitations
∙ There was no active reference in the study.
∙ As the study had a novel design, a number of design elements may have contributed to the study result.
Introduction
MDD is a recurrent, chronic, and seriously impairing disorder associated with substantial symptom severity (Reference Kessler, Berglund, Demler, Jin, Koretz and Merikangas1). Despite sufficient availability of different classes of antidepressants, most patients with MDD do not achieve adequate response or remission (Reference Han, Wang, Kato, Lee, Patkar, Masand and Pae2). Approximately 50% of patients with MDD do not achieve response to antidepressant treatment (Reference Papakostas3). In the STAR*D trial, one-third of patients with MDD did not achieve remission after as many as four different treatment strategies (4).
Treatment options for patients with inadequate response to ADT include switching to another ADT, adding a second antidepressant in combination, or adding another medication as adjunctive therapy (e.g. lithium, thyroid hormone, atypical antipsychotics, stimulants) (Reference Connolly and Thase5). Of these strategies, augmentation with an atypical antipsychotic is the most systematically and rigorously studied, and is supported by the strongest evidence base (Reference Connolly and Thase5,Reference Nelson and Papakostas6).
Brexpiprazole is a serotonin-dopamine activity modulator that acts as a partial agonist at the serotonin 5-HT1A and dopamine D2 receptors, and as an antagonist at the 5-HT2A and noradrenaline α1B/2C receptors, all with subnanomolar potency (7).
The efficacy and safety of brexpiprazole as adjunctive treatment to ADT over 6 weeks was demonstrated in two pivotal Phase 3, fixed-dose studies in MDD (Pyxis and Polaris) (8,9), and recently also in another fixed-dose study (Sirius) (Reference Hobart, Skuban, Zhang, Augustine, Brewer, Hefting, Sanchez and McQuade10) and a flexible-dose study (Delphinus) (11).
Brexpiprazole is approved in the United States and Saudi Arabia for use as adjunctive therapy to antidepressants for the treatment of MDD, and in the United States, Australia, Canada, Saudi Arabia, and Japan as monotherapy for treatment of schizophrenia.
Maintenance of efficacy is an important parameter in the successful treatment of MDD. However, studies of the maintenance of efficacy in patients with MDD treated with an adjunct second-generation antipsychotic are scarce (12,Reference Brunner, Tohen, Osuntokun, Landry and Thase13), with only one published study available at the time of initiation of the present study (12).
Aims of the study
The aim of the present study (Argo; NCT01838681; 2012-001380-76) was to evaluate brexpiprazole adjunctive to ADTs as maintenance treatment in patients with MDD with an inadequate response to ADT, utilising a novel, previously untried study design.
Methods
Study design and patients
This was a Phase 3, multicentre, randomised, double-blind, parallel-group, placebo-controlled, flexible-dose, long-term study, conducted at 112 sites across 16 countries in Asia (Republic of Korea), Europe (Bulgaria, Estonia, Finland, Germany, Latvia, Lithuania, Poland, Romania, Russia, Sweden, Ukraine, and United Kingdom), Latin America (Mexico), and North America (Canada, the USA). The study was designed and conducted in accordance with the principles of the Declaration of Helsinki and the study protocol and amendments were approved by the governing institutional review board or independent ethics committee for each investigational site or country, as appropriate.
The study included male and female outpatients, ≥18 and ≤75 years of age, with a primary diagnosis of MDD according to DSM-IV-TR® criteria (14) [current Major Depressive Episode (MDE) confirmed using the Mini-International Neuropsychiatric Interview (MINI) (15)], who had a Montgomery–Åsberg Depression Rating Scale (MADRS) (Reference Montgomery and Åsberg16) total score ≥26 at the screening visit and at the start of the prospective treatment period; had a Clinical Global Impression-Severity (CGI-S) (Reference Guy17) score ≥4 at the screening visit and at the start of the prospective treatment period; had had the current MDE for ≥8 weeks. Further, patients had an insufficient response to at least one and no more than three adequate ADTs (including the treatment a patient was taking at screening) for the current MDE; for the most recent antidepressant treatment, this had to be documented by self-report as <50% response on the Antidepressant Treatment Response Questionnaire (Reference Fava18). Exclusion criteria included a DSM-IV-TR® Axis I diagnosis other than MDD; presenting with suicidal ideation or behaviour; substance abuse or dependence within the past 180 days. All patients provided written informed consent before the start of the study.
The study consisted of a screening period and blinded treatment period (Fig. 1a). The blinded treatment period consisted of an 8-week prospective treatment period and a 24-week randomised, double-blind treatment period. The study design was blinded to the investigators and the patients (Fig. 1b) in order to prevent rater and patient expectations influencing the objective assessment of the symptom severity. Neither investigator nor patient knew when the randomised adjunctive treatment started or what the randomisation criteria were. Following the blinded treatment period there was a safety follow-up comprising a telephone contact or a clinic visit 30 (±3) days after last dose of study medication.
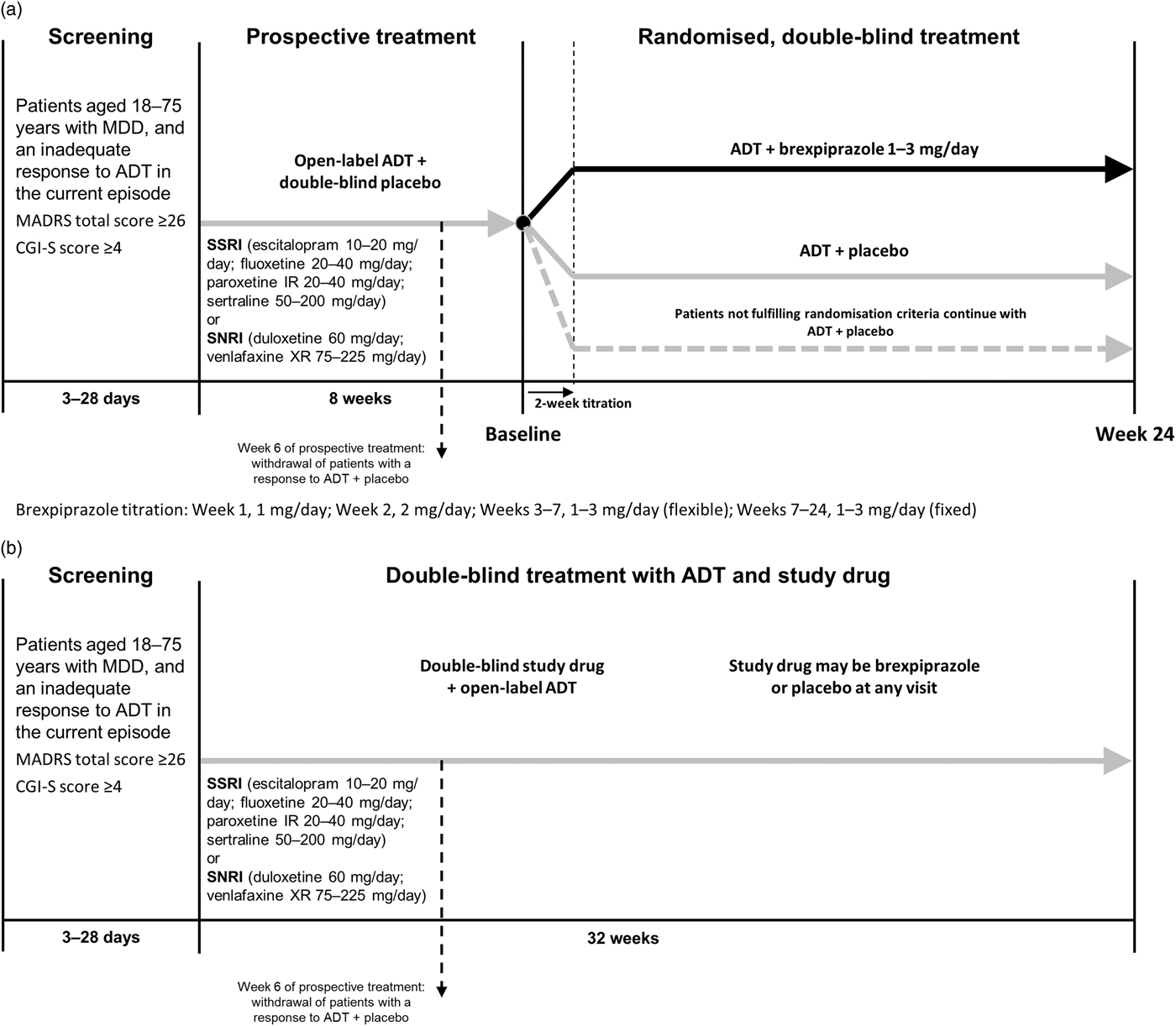
Fig. 1 Study design. (a) Unblinded study design. (b) Blinded study design. ADT, antidepressant therapy; CGI-S, Clinical Global Impression-Severity; IR, immediate release; MADRS, Montgomery–Åsberg Depression Rating Scale; MDD, major depressive disorder; SNRI, serotonin–noradrenaline reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; XR, extended release.
The study protocol was amended during the study and the major amendment concerned: removal of one part of the initial definition of inadequate response (CGI-S score ≥4 at every visit in the prospective treatment period) to more closely match the criteria used in the pivotal acute treatment Phase 3 studies; withdrawal of patients with early response to ADT at Week 6; removal of randomised time of randomisation (after 8 or 10 weeks in the prospective treatment period), thus having all patients fulfilling randomisation criteria randomised after 8 weeks in the prospective treatment period, and all patients ending double-blind treatment after 24 weeks. The methodology presented herein reflects the protocol after implementation of this protocol amendment as the majority of patients (>80%) were randomised according to this amendment. All randomised patients were included in the analyses according to the analysis set definition.
Patients received 8 weeks of open-label treatment with one of six ADTs and double-blind placebo treatment (weekly visits the first 2 weeks; biweekly weeks 2–8) during the prospective treatment period. The dose of the ADT was titrated during the first 4 weeks to increase the likelihood of ADT success (online Supplementary Appendix Table S1). The investigator could adjust the ADT dose based on tolerability during the first 4 weeks of the prospective treatment period; after Week 4, the dose had to remain stable.
Patients who achieved response [defined as a decrease of ≥50% in MADRS total score since the start of the prospective treatment period or a Clinical Global Impression-Improvement (CGI-I) score <3] at any of the visits during the first 6 weeks of the prospective treatment period were withdrawn from the study after 6 weeks in the prospective treatment period.
Patients not fulfilling response criteria continued in the prospective treatment period. Patients who continued after 6 weeks in the prospective treatment period, but did not fulfil the randomisation criteria after 8 weeks of prospective treatment, continued with the same treatment they received in the prospective treatment period (ADT+placebo) and followed the same assessment schedule as the randomised patients in the double-blind treatment period.
Patients with inadequate response to ADT in the prospective treatment period were randomised to receive 24 weeks (biweekly visits baseline to Week 10 of the randomised period; monthly visits Weeks 10–22; final visit at Week 24) of double-blind treatment with 1–3 mg/day brexpiprazole or placebo (1 : 1) in addition to the open-label ADT they received in the prospective treatment period. Patients were eligible for randomisation if they met all of the following criteria:
∙ <50% reduction in MADRS total score between the start of prospective treatment and all visits in the prospective period
∙ MADRS total score ≥18 at the randomisation visit
∙ CGI-I score ≥3 (minimally improved) at every visit in the prospective period.
Patients in the brexpiprazole group received 1 mg/day in Week 1 followed by 2 mg/day in Week 2. From Week 3 onwards, the dose was 3 mg/day for the remainder of the study. The dose of brexpiprazole could be adjusted in a blinded way by requesting a dose decrease or dose increase during Weeks 3–7 to optimise the clinical effect and tolerability. This could result in a brexpiprazole dose of 1, 2, or 3 mg/day. From Week 7 onwards, the dose had to be stable (online Supplementary Appendix Table S2).
Randomisation and masking
At each visit, the MADRS total score and the CGI-S and CGI-I scores were entered into, and analysed by, an interactive voice/web-response system (IVRS/IWRS) in order to maintain the blinding. Treatment assignments were based on a computer-generated randomisation code provided by the study sponsor. Patients were randomised in a 1 : 1 ratio to brexpiprazole or placebo. Double-blind study medications were provided by the sponsor, in 2-week wallet cards, and were assigned by the IVRS/IWRS. The tablets for brexpiprazole and placebo were identical in appearance.
The investigator was only to break the randomisation code if knowledge of the study medication was necessary to provide optimal treatment to the patient in an emergency situation. During the prospective treatment period, the code was broken for one patient because of a haemorrhagic stroke reported as a serious adverse event (SAE).
Endpoints
The primary endpoint was full remission, defined as a MADRS total score ≤10 and a ≥50% decrease from randomisation (i.e. baseline) in MADRS total score for at least 8 consecutive weeks during randomised treatment. Key secondary endpoints were full functional remission, defined as a Sheehan Disability Scale (SDS) (Reference Sheehan, Harnett-Sheehan and Raj19) total score ≤6 and SDS domain scores ≤2, for at least 8 consecutive weeks during randomised treatment; and full global score remission, defined as a CGI-S score ≤2 for at least 8 consecutive weeks during randomised treatment. Secondary endpoints included change from baseline in MADRS total score after 6 and 24 weeks of randomised treatment.
Safety was assessed by spontaneous reporting of adverse events (AEs), clinical laboratory tests, physical examination, vital signs, body mass index, and electrocardiograms (ECGs). Extrapyramidal symptoms (EPS) were formally assessed using the Simpson–Angus Scale (SAS) (Reference Simpson and Angus20), Barnes Akathisia Rating Scale (BARS) (Reference Barnes21), and Abnormal Involuntary Movement Scale (AIMS) (Reference Guy22). Suicidality was assessed using the electronic Columbia-Suicide Severity Rating Scale (C-SSRS) (23).
Statistical methods
When 440 patients had been randomised, a blinded data review was performed and the joint rate of full remission was estimated to be lower than originally predicted; the sample size therefore had to be adjusted. The original odds ratio (OR) of 1.62 was used to translate the assumed treatment effect. Assuming an average full remission rate of 20% and an advantage difference of 7.6% between ADT+brexpiprazole and ADT+placebo, 434 randomised patients per treatment arm were needed to provide a power of 80%.
Efficacy and safety analyses were based on all randomised patients who took at least one dose of randomised study medication in the randomised, double-blind treatment period. The primary efficacy analysis used a logistic regression (LREG) model including MADRS total score at baseline as a covariate and treatment group, country, and randomisation criteria as fixed effects. Three sets of randomisation criteria were applied: randomised after 8 weeks using the original criteria [ORG (Week 8)]; randomised after 10 weeks using the original criteria [ORG (Week 10)]; and randomised after 8 weeks using the amended criteria [AMEND (Week 8)]. The key secondary endpoints were analysed using the LREG model described for the primary endpoint, but with the MADRS total score replaced with the SDS total score in the analysis of full functional remission, and with the CGI-S score in the analysis of full global score remission. The continuous secondary endpoints measured at several visits were analysed using a mixed model for repeated measurements with treatment and country as fixed effects and the relevant score at randomisation as a covariate. In addition, the randomisation criteria were included as a fixed effect and as interacting with time since randomisation. Interaction between time since randomisation and treatment was included as a fixed effect, and interaction between time since randomisation and value at randomisation was included as a covariate. The covariance within patients was modelled using an unstructured variance-covariance matrix.
The randomisation visit was used as baseline assessment in the randomised, double-blind treatment period. If there was no assessment at the randomisation visit for the safety endpoints, the assessment from the last visit before the randomisation visit was used as baseline assessment.
For the ADT+brexpiprazole and ADT+placebo comparisons, the following sequence of hierarchically ordered primary and key secondary endpoints was applied:
∙ full remission
∙ full functional remission
∙ full global score remission.
The overall significance level was 0.05. Only if the primary endpoint was statistically significant would confirmatory testing continue with the key secondary endpoints.
Results
Patients
The study was initiated on 28 May 2013 and completed on 8 June 2016. A total of 2517 patients were screened, and 1986 entered the prospective treatment period (Fig. 2). A total of 1661 (83.8%) patients completed the prospective treatment period; 775 (39.1%) patients demonstrated response to ADT+placebo and were not eligible for randomisation, while 886 (44.7%) patients demonstrated inadequate response to ADT+placebo and were therefore eligible for randomisation. Inadequate responders continued on the same ADT and were randomised to adjunctive brexpiprazole 1–3 mg/day (n=444) or adjunctive placebo (n=442). A total of 444 and 441 patients received at least one dose of brexpiprazole or placebo, respectively, in the randomised period and formed the analysis population. Of the randomised patients, 349 (78.6%) receiving ADT+brexpiprazole and 380 (86.0%) receiving ADT+placebo completed the randomised treatment phase. In both treatment groups, the most common primary reasons for withdrawal were withdrawal of consent (7.1%), AEs (4.5%), or lack of efficacy (2.3%).

Fig. 2 Patient flow chart. ADT, antidepressant therapy; APTS, all patients treated set; FAS, full analysis set.
Baseline demographic and clinical characteristics were similar between the treatment groups (Table 1).
Table 1 Demographics and clinical characteristics
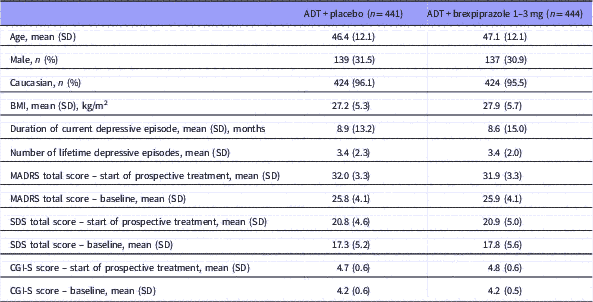
ADT, antidepressant therapy; BMI, body mass index; CGI-S, Clinical Global Impression-Severity; MADRS, Montgomery–Åsberg Depression Rating Scale; MDD, major depressive disorder; SDS, Sheehan Disability Scale.
The average modal and mean doses of brexpiprazole were 2.83 mg/day (n=442) and 2.69 mg/day (n=444), respectively. At the last visit 88% of the patients in the brexpiprazole group received 3 mg/day brexpiprazole.
Efficacy
The primary efficacy analysis of full remission failed to show a statistically significant difference between ADT+brexpiprazole and ADT+placebo. The proportion of patients who achieved full remission was 21.4% in the ADT+brexpiprazole group and 24.9% in the ADT+placebo group (OR: 0.83; p=0.2641) (Table 2). The key secondary efficacy analyses of full functional remission and full global score remission also failed to demonstrate an advantage of brexpiprazole over placebo (Table 2). The secondary analysis of change from baseline in MADRS total score after randomised treatment showed no difference between ADT+brexpiprazole and ADT+placebo in mean change from baseline in MADRS total score at Week 6 [−0.4 (95% confidence interval −1.2; 0.4), p=0.3259] (Fig. 3).

Fig. 3 Mean change from baseline in MADRS total score. ADT, antidepressant therapy; LS, least squares; MADRS, Montgomery–Åsberg Depression Rating Scale; MMRM, mixed model for repeated measurements; SE, standard error.
Table 2 Primary and key secondary endpoints

ADT, antidepressant therapy; CI, confidence interval; CGI-S, Clinical Global Impression-Severity; MADRS, Montgomery–Åsberg Depression Rating Scale; SDS, Sheehan Disability Scale.
Logistic regression model with treatment, design factor D and pooled country as fixed effects and MADRS total score/SDS total score/CGI-S score at baseline as covariate.
* MADRS total score ≤10 and ≥50% decrease from baseline observed for ≥8 consecutive weeks during the randomised treatment period.
† SDS total score ≤6 and all SDS domain scores ≤2 observed for ≥8 consecutive weeks during the randomised treatment period. If work/school item was missing it was imputed as the average of the social life/leisure activities and home/family responsibilities scores before the full functional remission was derived.
‡ CGI-S score ≤2 observed for ≥8 consecutive weeks during the randomised treatment period.
Safety and tolerability
The most frequent treatment-emergent AEs (TEAEs) (≥5%) in the randomised treatment period in patients receiving ADT+brexpiprazole were weight increased (9.5% vs. 5.0% in ADT+placebo), headache (7.7% vs. 7.0% in ADT+placebo), nasopharyngitis (6.3% vs. 7.7% in ADT+placebo), and accidental overdose [6.1% vs. 5.7% in ADT+placebo; reported as TEAE if >1 tablet of study medication (including ADTs) had been taken] (Table 3). The majority of patients with TEAEs had TEAEs that were either mild or moderate; the overall incidence of severe TEAEs was 6% in the ADT+brexpiprazole group and 5% in the ADT+placebo group.
Table 3 Treatment-emergent adverse events (TEAEs)

ADT, antidepressant therapy.
Values are n (%).
The incidence of TEAEs leading to withdrawal during the randomised treatment period was 6.3% in the ADT+brexpiprazole group and 3.4% in the ADT+placebo group. The TEAEs leading to withdrawal in ≥2 patients in the ADT+brexpiprazole group were weight increased (three patients), akathisia, anxiety, depression, dyskinesia, fatigue, and suicidal ideation (each two patients).
In the randomised treatment period, SAEs were reported for nine patients (2.0%) in the ADT+brexpiprazole group and 13 patients (2.9%) in the ADT+placebo group. Two SAEs were reported by >1 patient in at least one treatment group: suicidal ideation – 2 patients (0.5%) in the ADT+brexpiprazole group and 3 patients (0.7%) in the ADT+placebo group; intentional overdose – 0 patients (0.0%) in the ADT+brexpiprazole group and 2 patients (0.5%) in the ADT+placebo group.
Two patients died during the study. 1: A woman suffered from a haemorrhagic stroke that occurred during the prospective treatment period after 43 days on ADT (fluoxetine)+placebo. There were no clinically relevant medical history or laboratory test findings. The patient died 10 days later; the SAE was considered not related to treatment by the investigator. 2: A man committed suicide by an intentional overdose of ‘unknown drug and alcohol’ after 136 days (non-randomised patient) on ADT (duloxetine)+placebo. The SAE was considered not related to treatment by the investigator. Neither patient received brexpiprazole at any time during the study.
Activating side effects were infrequently reported (akathisia, 4.7% vs. 0.9%; restlessness, 4.1% vs. 0.5%; insomnia, 1.8% vs. 0.9%; anxiety, 1.6% vs. 0.9%, and agitation 0.7% vs. 0% for ADT+brexpiprazole and ADT+placebo, respectively). Sedating side effects were also relatively uncommon (fatigue, 3.8% vs. 1.4%; somnolence, 2.9% vs. 1.4%, and sedation, 0% vs. 0%).
The proportion of patients with EPS-related TEAEs was 9.2% in the ADT+brexpiprazole group and 3.6% in the ADT+placebo group. The EPS-related TEAEs with an incidence ≥2% in either treatment group were (brexpiprazole vs. placebo): akathisia (4.7% vs. 0.9%) and tremor (2.9% vs. 2.0%). There were minor fluctuations in the mean (SD) SAS total scores, BARS global clinical assessment of akathisia scores, and AIMS total scores in both treatment groups during the randomised treatment period, with no clinically relevant differences between the treatment groups; SAS total scores at Week 24: ADT+brexpiprazole: 0.18 (0.68), ADT+placebo: 0.08 (0.41); BARS global scores at Week 24: ADT+brexpiprazole: 0.04 (0.20), ADT+placebo: 0.01 (0.07); AIMS total scores at Week 24: ADT+brexpiprazole: 0.06 (0.32), ADT+placebo: 0.02 (0.17).
In the ADT+brexpiprazole group, the median prolactin levels increased from baseline to Week 24 (Table 4); the median change from baseline in prolactin value peaked at Week 4 in both men (1.77 ng/ml) and women (7.68 ng/ml) and then decreased to Week 24. Post-baseline elevated prolactin values (>3 times upper limit of normal) were noted for 1/132 men (0.8%) and 8/305 women (2.6%) in the ADT+brexpiprazole group and 0 men (0%) and 2/298 women (0.7%) in the ADT+placebo group.
Table 4 Laboratory assessments – changes from baseline to Week 24

ADT, antidepressant therapy; HDL, high density lipoprotein; LDL, low density lipoprotein.
* Shift from baseline to any time post-baseline.
Mean changes in fasting glucose and lipid parameters were small (Table 4).
Mean (SD) weight gain from baseline to Week 24 was 2.1 (4.2) kg in the ADT+brexpiprazole group and 0.8 (3.3) kg in the ADT+placebo group; the increase in the ADT+brexpiprazole group mainly occurred during the first 10 weeks [1.7 (2.8) kg]. A larger proportion of patients in the ADT+brexpiprazole group (84/441, 19%) than in the ADT+placebo group (36/436, 8%) had a ≥7% weight increase from baseline.
No meaningful differences between the ADT+brexpiprazole group and the ADT+placebo group were seen in ECG parameters and vital signs.
Administration of brexpiprazole with ADTs did not appear to increase suicidal behaviour or ideation. Three patients (0.7%) in each treatment group had suicidal behaviour. Treatment-emergent suicidal ideation was reported by 35 patients (8.0%) in the ADT+placebo group and by 34 patients (7.7%) in the ADT+brexpiprazole group.
Discussion
In this flexible-dose, 24-week study, adjunctive brexpiprazole as maintenance treatment failed to differentiate from ADT+ placebo on the primary endpoint – full remission. The key secondary endpoints of full functional remission and full global score remission also did not show an advantage of adjunctive brexpiprazole over ADT+placebo.
When the current study was initiated there was only one published study available assessing the maintenance of efficacy in patients with MDD treated with an adjunct second-generation antipsychotic (12). In that study, the primary endpoint was time to relapse for patients treated with the combination of citalopram+risperidone versus citalopram+placebo, investigated using the commonly applied randomised withdrawal study design. The study was negative, which prompted the novel design and primary endpoint of the current study, which had never been tried before.
Negative studies are common in MDD (Reference Schatzberg24), even in medications that have been proven to work. One unique reason cannot be identified to explain the results of the current study. When analysing the secondary endpoint of change from baseline in MADRS total score, adjunctive brexpiprazole did not separate from placebo at Week 6. This finding stands in contrast to what has been observed in four previous studies with brexpiprazole (8–11). In this respect, it is of interest that the patient population in the current study differs from those of the previous trials, having a much shorter duration of the current depressive episode.
The fact that the results from previous short-term studies could not be replicated may suggest that no conclusions should be drawn about the maintenance of efficacy of adjunctive treatment with brexpiprazole based on the current study.
The Delphinus study (11) utilised a similar blinded study design as the current study, and despite the former being a 6-week short-term study, some interesting comparisons can be made.
The original blinded study design of the current study, where investigators and site staff were blinded to the treatment periods, the randomisation criteria and the timing of randomisation, allowed biases to be minimised. With the amendment, a Week 6 withdrawal visit was introduced to the prospective treatment period in order to discontinue early responders. Although only 148 patients were withdrawn due to early response at this withdrawal visit, with this addition investigator bias might have been introduced, at least with regard to the existence of a prospective treatment period without brexpiprazole.
The proportion of responders to prospective treatment is much smaller than that seen in the Delphinus study (11), and consequently, a much larger proportion of ADT+placebo patients (44.7%) were randomised in the current study than in Delphinus (23.1%). It is likely that the introduction of the withdrawal visit after 6 weeks of prospective treatment is a contributory factor to this difference in the randomisation rate.
The study had a high completion rate in both treatment arms, with a completion rate in the ADT+brexpiprazole group of 78.6% and 86.0% in the ADT+placebo group, which is remarkably high for a long-term study (12,Reference Brunner, Tohen, Osuntokun, Landry and Thase13,Reference Ball, Atkinson, Sparks, Bangs, Goldberger and Dube25–Reference Nelson, Skuban, Hobart, Weiss, Weiller and Thase27), although direct comparisons with open-label studies, primarily designed to assess long-term safety, or double-blind relapse-prevention studies with protocol-specified withdrawal criteria, are not straight-forward. Nevertheless, the completion rate approached those of the 6-week short-term studies (8–11), highlighting the ability of the study investigators to retain patients in the study. The withdrawal rate due to TEAEs was somewhat higher than in the short-term studies (8–11), but still indicates that adjunctive brexpiprazole was well tolerated.
The most commonly reported TEAE in the ADT+brexpiprazole group was weight increased (9.5%), which is reported here at a slightly higher incidence than in the short-term (6 weeks) studies (8–11), as expected, but at a much lower incidence than in the 52-week open-label studies (25.5%) (Reference Nelson, Skuban, Hobart, Weiss, Weiller and Thase27). Weight gain in the ADT+brexpiprazole group was 2.1 kg after 24 weeks, again slightly higher than in the short-term studies (8–11), but lower than in the 52-week open-label studies (3.2 kg) (Reference Nelson, Skuban, Hobart, Weiss, Weiller and Thase27). It should also be noted that the patients on placebo gained 0.8 kg in weight. Overall, treatment with brexpiprazole (1–3 mg/day) for 24 weeks was safe and well tolerated in this patient population with MDD.
In addition to the potential drawbacks with a number of design elements in the current study that already have been mentioned, one limitation that warrants mentioning is the lack of an active reference.
In conclusion, adjunctive brexpiprazole as maintenance treatment did not differentiate from ADT+placebo on the primary endpoint of full remission. The fact that the results from previous short-term studies could not be replicated may suggest that no conclusions should be drawn about the maintenance of efficacy of adjunctive treatment with brexpiprazole based on the current study. A number of design elements in this previously untried study design may have contributed to the study result. As in the previous studies, brexpiprazole was well tolerated in patients with MDD when administered as adjunctive therapy for 24 weeks, with no unexpected side effects.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/neu.2018.23
Acknowledgements
Under the direction of the authors, Johan Hellsten, PhD (a full-time employee of H. Lundbeck A/S) drafted the initial version of the manuscript, and edited subsequent versions. The authors are entirely responsible for the scientific content of this paper.
Financial Support
This work was supported by Otsuka Pharmaceutical Development & Commercialization Inc. (Princeton, NJ, USA) and H. Lundbeck A/S, Valby, Denmark.
Conflicts of Interest
In the past 3 years, M.B. is/has been a consultant for Allergan, Aristo, Boehringer-Ingelheim, Janssen, Lundbeck, neuraxpharm, Otsuka, Servier, and has received speaker honoraria from AstraZeneca, GlaxoSmithKline, Lilly, Otsuka, Pfizer and Servier. N.H., A.L., and M.K.J. are full-time employees of H. Lundbeck A/S. M.H. is a full-time employee of Otsuka Pharmaceutical Development & Commercialization Inc.
Ethical Standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.









