Introduction
Strongyloides is a genus containing some 50 species of obligate gastrointestinal parasites of vertebrates (mammals, birds, reptiles and amphibians) (Speare, Reference Speare and Grove1989). Two species infect humans: S. stercoralis and S. fuelleborni-like forms (including S. fuelleborni kellyi). The former has a cosmopolitan distribution in tropical and subtropical regions (Schad, Reference Schad and Grove1989) and the latter naturally infects non-human primates in Africa but can also infect humans there (Ashford et al., Reference Ashford, Barnish and Viney1992). Human infection with S. fuelleborni was first recorded in Zimbabwe (Grove, Reference Grove and Grove1989), and it subsequently proved to be distributed widely in sub-Saharan countries (e.g. Central African Republic, Cameroon and Ethiopia) (Pampiglione & Ricciardi, Reference Pampiglione and Ricciardi1971; Hasegawa et al., Reference Hasegawa2010) and South-East Asia, including Thailand (Thanchomnang et al., Reference Thanchomnang2017). This parasite is common among Old-World primates, including rhesus monkeys and macaques (Sandground, Reference Sandground1925). In addition, S. fuelleborni kellyi has been reported from humans in Papua New Guinea (Ashford et al., Reference Ashford, Barnish and Viney1992).
The long-tailed macaque (Macaca fascicularis) is a species of Old-World monkey that is widely distributed in tropical South-East Asia (Fooden, Reference Fooden1995; Malaivijitnond & Hamada, Reference Malaivijitnond and Hamada2008). Its natural range extends southwards and eastwards from south-eastern Bangladesh and Myanmar, through the southern part of the Indochinese Peninsula (Thailand, Cambodia, Lao PDR and Vietnam) and into Malaysia, Singapore, and the islands of Sumatra, Borneo, Java and the Philippines (Eudey, Reference Eudey2008). In Thailand, the long-tailed macaque is distributed extensively, from the lower northern region to the southern part of the country (Malaivijitnond & Hamada, Reference Malaivijitnond and Hamada2008). Because of human intervention, the habitats of the long-tailed macaque have been greatly altered from natural forests to temple grounds or park areas close to human settlements. The macaque's natural foraging behaviour has also changed, towards begging and searching garbage for food. The possibility of zoonotic transmission of animal parasites from this primate to humans is a real concern. Therefore, we have examined faecal samples of long-tailed macaques inhabiting some tourist attraction sites in Thailand and Lao PDR in a search for Strongyloides. Recently, S. fuelleborni was recorded and identified, based on morphological criteria, in long-tailed macaques from north-east Thailand (Wenz-Mücke et al., Reference Wenz-Mücke2013) and there has been a single report of human infection in the same region (Thanchomnang et al., Reference Thanchomnang2017). However, there have been no previous attempts at molecular identification of S. fuelleborni in monkeys in Thailand and Lao PDR. Here, we report amplification and sequencing of a portion of the 18S rRNA and cox1 regions and use the sequences for identification of the Strongyloides species present in long-tailed macaques.
Materials and methods
Study areas and populations
Fresh faecal droppings from 243 individual macaques were collected in January and February 2018. Small groups of macaques were followed during their complete daily activity period (06.00–12.00). Individual faecal samples were collected directly after excretion. Faecal samples were collected at Kumphawapi Monkey Park, Kumphawapi District, Udon Thani Province (17°06′41.8″N, 103°01′03.4″E) (n = 67) and Kosumpee Forest Park, Kosumpisai District, Maha Sarakham Province (16°15′190″N, 103°04′024″E) (n = 110) in north-eastern Thailand, and in Champon District, Savannakhet Province, Lao PDR (16°32.608′N, 105°15.081'E) (n = 66). A map showing these localities is provided in fig. 1.

Fig. 1. Map of the study areas in Thailand and Lao PDR. Numbers and shading indicate study province in each country.
Stool examination and sample collection
Strongyloides free-living adults and larvae were detected using an agar plate culture (Koga et al., Reference Koga1991). A 3 g portion of each faecal sample (n = 243) was placed on a nutrient agar plate in the field. Strongyloides rhabditiform larvae and free-living adults were observed and collected after 3 days of culture at room temperature. Parasite stages harvested were fixed in 70% ethanol, transported to the laboratory, and kept at −20°C until used for molecular identification.
DNA extraction, polymerase chain reaction (PCR), and sequencing
A single free-living adult male worm was taken from each positive culture, individually crushed with a disposable polypropylene pestle (Bellco Glass, Vineland, NJ, USA) and DNA extracted using the Nucleospin Tissue Kit (Macherey-Nagel GmbH & Co, Duren, Germany). Extracted DNA was eluted in 50 μl of elution buffer, 5 μl of which was used for amplification. Primers specific for portions of the Strongyloides 18S rRNA gene and of the mitochondrial cox1 gene were used as previously described (Laymanivong et al., Reference Laymanivong2016). PCR cycling conditions were also as reported in Laymanivong et al. (Reference Laymanivong2016). DNA direct sequencing was done using the Applied Biosystems 3730×I DNA Analyzer and ABI Big Dye Version 3.1 (Applied Biosystems, Foster City, CA, USA) in both directions, using the PCR primers as sequencing primers. The gene sequences were analysed using a nucleotide BLAST search via NCBI. New sequences of both regions were aligned with reference sequences from the NCBI. The cox1 alignment was 646 bp in length. Two different alignments were analysed for the partial 18S gene. The longer one was 356 bp after trimming the primer sequences and reducing the alignment to the length of the shortest published sequence retained in the analysis. A shorter alignment of 219 bp permitted inclusion of additional sequences, primarily from Africa, which overlapped with our sequences by that amount. BioEdit (Hall, Reference Hall1999) was used for preparing the alignments.
Alignment and phylogenetic analysis
The 18S rRNA sequences of Strongyloides spp. were aligned with reference sequences from the NCBI database using BioEdit software (Hall, Reference Hall1999). Bayesian inference (MrBayes v3.2; Ronquist et al., Reference Ronquist2012) was used to construct a phylogenetic tree from the 18S rRNA alignment. Maximum likelihood (ML) and neighbour-joining (NJ) methods, as implemented in MEGA7 (Kumar et al., Reference Kumar, Stecher and Tamura2016), were also used. The substitution models for both datasets were chosen using the Bayesian information criterion (BIC) in MEGA7 software: the lowest BIC score is considered to best describe the substitution pattern. For the 18S rRNA alignment, the HKY + G model was selected. MrBayes was run for 500,000 generations, by which time the standard deviation of split frequencies had fallen below 0.01, and trees sampled every 300 generations. In all Bayesian analyses, consensus trees were generated using the command “con type = half compat”, which results in a 50 majority-rule tree. For the maximum-likelihood and neighbour-joining methods, the Kimura two-parameter (K2) model was used (Kimura, Reference Kimura1980). Branch support in ML and NJ was assessed by bootstrapping with 1000 replications (Kumar et al., Reference Kumar, Stecher and Tamura2016). DnaSP v5 was used to infer haplotype composition and haplotype diversity (Hd) (Librado & Rozas, Reference Librado and Rozas2009). A median-joining network was constructed using the Network software version 5.0.0.0 (Fluxus Technology Ltd., www.fluxus-engeneering.com). All sequences have been deposited in the GenBank database (table 1).
Table 1. Strongyloides fuelleborni sequences obtained and analysed in the present study.

Results
In total, 243 faecal samples were collected from free-ranging long-tailed macaques inhabiting three localities in Thailand and Lao PDR. Ninety-six of these (39.5%) were positive for Strongyloides by agar plate culture. The prevalence in Thailand was 31.1% (55/177) and in Lao PDR it was 62.1% (41/66). From each positive culture, one adult male free-living worm was used for DNA extraction, amplification and sequencing. The DNA of Strongyloides sp. samples was detected with PCR targeting the 18S rRNA and cox1 genes. Each of the 96 adult male worms possessed an 18S sequence identical to GenBank accessions of S. fuelleborni (100% similarity and 100% query coverage; KY081222, AB272235) as reported in studies from Japan and Thailand (fig. 2a). In the tree inferred from the shorter 18S alignment, which permitted addition of partial sequences from Africa, some African sequences were identical to those from Asia, and some were very slightly different, causing them to group in a second clade (fig. 2b).

Fig. 2. Maximum-likelihood phylogenetic tree of Strongyloides spp. based on partial 18S rRNA sequences. Support values (Bayesian posterior probabilities/ML bootstrap/NJ bootstrap) are shown above the branches. A dash (−) instead of a numerical support value indicates that a particular grouping was not found by that method of analysis. Bold letters indicate sequences obtained in the present study. (a) Phylogenetic tree inferred from a 356-bp alignment of Strongyloides fuelleborni from Thailand, Lao PDR and Japan, and (b) a tree inferred from a 219-bp alignment which permitted addition of S. fuelleborni sequences from Central African Republic, Gabon, Uganda and Tanzania, which overlap with our sequences by that amount. Sequence AB453320 derived from sample of Japanese researcher, acquired in Tanzania and diagnosed in Japan.
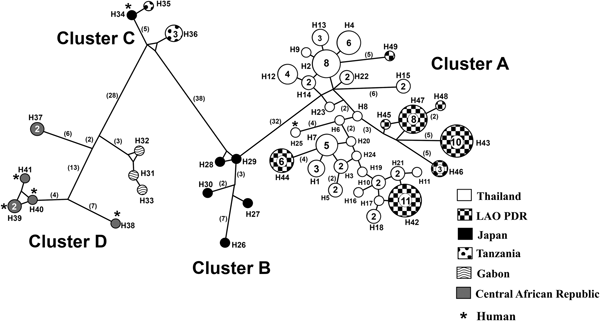
Similarly, all 96 cox1 sequences obtained by us showed 98–99% identity with cox1 sequences of S. fuelleborni from humans in Thailand (GenBank accession no. KY081233, 100% query coverage) and 93–94% similarity with sequences of this species from macaques (Macaca fuscata fuscata) in Japan (AB526290, AB526291, AB526292, AB526293 and AB526294: 90–91% coverage). Our 96 sequences represented 32 haplotypes, all new. Phylogenetic analysis of cox1 sequences placed S. fuelleborni and S. stercoralis in separate clades (100% bootstrap values, data not shown). Strongyloides fuelleborni sequences from Thailand and Lao PDR were closely related to those from Japan, but located in separate clades from those from Tanzania, Gabon and Central African Republic. When other sequences from GenBank were included (table 2), a median-joining network clustered the sequences into four groups (A, B, C and D), with a total of 49 haplotypes (total Hd = 0.9670; Hd from Thailand and Lao PDR = 0.953; Hd from Japan = 1; Hd from Tanzania = 0.7, and Hd from Gabon and Central African Republic = 0.956) (fig. 3). Cluster A consisted of our 32 new haplotypes from Thailand and Lao PDR, together with haplotype 25, a human isolate recovered in Thailand (Thanchomnang et al., Reference Thanchomnang2017). Cluster B consisted of five haplotypes from Japan (macaques). Cluster C consisted of three haplotypes from Tanzania: haplotypes 35 and 36 were from chimpanzees and yellow baboons, respectively, and haplotype 34 was from a Japanese researcher working with wild chimpanzees in the area. Cluster D consisted of three haplotypes from Gabon (chimpanzee and gorilla) and five haplotypes from Central African Republic (humans, chimpanzees and gorillas).

Fig. 3. Median-joining network of cox1 sequences of Strongyloides fuelleborni. The shading patterns indicate the different geographical sources. Numbers in parentheses indicate numbers of nucleotide changes inferred as having occurred between nodes. Numbers in circles indicate number of samples with that haplotype, when this is >1. Cluster A consists of haplotypes 1–24 from macaques (from the present study), haplotype 25 from a human in Thailand (Thanchomnang et al. Reference Thanchomnang2017), and haplotypes 42–49 from macaques, collected from Lao PDR (from the present study). Cluster B consists of haplotypes 26–30 from macaques, collected from Japan. Cluster C consists of haplotype 34 from a Japanese researcher who acquired the infection while working with wild chimpanzees in Tanzania, and haplotypes 35–36 from chimpanzees and yellow baboons in Tanzania. Cluster D consists of haplotypes 31–33 from chimpanzees and gorillas, collected from Gabon, haplotype 37 from chimpanzee and gorilla and haplotypes 38–41 from humans, collected from Central African Republic.
Table 2. GenBank accession numbers of previously published Strongyloides fuelleborni cox1 sequences used to construct the haplotype network.

Discussion
Molecular methods for discriminating among Strongyloides spp. are important in situations in which the parasite morphology and epidemiological information are similar (Hasegawa et al., Reference Hasegawa2010, Reference Hasegawa2016; Thanchomnang et al., Reference Thanchomnang2017). Here, we have presented the first molecular identification of S. fuelleborni infections in long-tailed macaques in Thailand and Lao PDR. Sequences from a variable region of the 18S rRNA gene were used to identify Strongyloides worms to species. A portion of the cox1 gene was used to examine genetic diversity within S. fuelleborni. Phylogenetic analysis of the 18S rRNA gene can distinguish S. fuelleborni from other species of Strongyloides (fig. 2). All of the 18S rRNA nucleotide sequences of S. fuelleborni from 96 obtained samples were identical with published S. fuelleborni sequences from Japan and Thailand. Addition of 18S sequences from Africa in a shorter alignment revealed some African sequences identical with those from Asia and some in a different, but closely related, clade. The 18S gene is known to be highly conserved within species.
However, we found 49 haplotypes among cox1 sequences from S. fuelleborni when sequences from GenBank were included in the analysis. Thirty-three haplotypes were from Thailand and Lao PDR, six from Japan, two from Tanzania, three from Gabon and five from Central African Republic. The haplotypes fell into four separate clusters, each containing examples from humans, except cluster B (macaques in Japan). There was a strong geographical structure among clusters (Hasegawa et al., Reference Hasegawa2016). Clearly, our sampling efforts in Thailand and Lao PDR have been more extensive than in previous studies in Japan (Hasegawa et al., Reference Hasegawa2010) and various parts of Africa (Hasegawa et al., Reference Hasegawa2016). Hasegawa et al. (Reference Hasegawa2010) obtained a Strongyloides sample from a Japanese mammalogist, who had participated in a field survey in Tanzania. A sequence of the cox1 gene from this case was almost identical to those of worms in Tanzanian chimpanzees and yellow baboons. The same authors noted that sequences available at the time (n = 24) could be divided into three phylogenetic groups, which corresponded to geographical localities (Japan, Tanzania and Gabon) but not to host species. A later analysis by Hasegawa et al. (Reference Hasegawa2016) included 14 additional cox1 sequences of Strongyloides larvae obtained from humans, lowland gorillas and chimpanzees inhabiting Central African Republic, and chimpanzees living in Uganda. The haplotypes fell clearly into two lineages, corresponding to S. stercoralis and S. fuelleborni. Cox1 haplotypes of S. fuelleborni formed clades according to their geographical locality. The S. fuelleborni haplotypes from the humans and apes in Central African Republic formed separate clades, closer to those from Ugandan chimpanzees and Gabonese apes, respectively. When aligned with our new sequences, the same pattern was found on a broader scale: geography, rather than host species, dictates the pattern of clustering. The high diversity reported by Hasegawa et al. (Reference Hasegawa2016) is matched by the diversity of sequences in our study that the haplotypes of S. fuelleborni formed clades according to their geographical locality.
Despite the geographical clustering evident in our cox1 haplotype network, we cannot infer much about the biogeography of S. fuelleborni. The Thai/Lao cluster is closest to the cluster from Japan, but the branch connecting these two clusters is almost as long as the branch connecting the Japanese cluster with African clusters. The long branches separating African sequences suggest that additional sampling there will reveal many additional haplotypes. Primate populations in large parts of Asia also remain to be sampled.
Several molecular methods have been developed for the identification of Strongyloides spp. For example, ITS1 and 18S rRNA have been used for PCR-based identification of S. fuelleborni and S. stercoralis in orangutans and in humans working with wild orangutans in Central and East Kalimantan, Borneo, Indonesia (Labes et al., Reference Labes2011), 18S rDNA and cox1 genes were used for identifying S. fuelleborni and S. stercoralis in humans, several monkey species and dogs (Hasegawa et al., Reference Hasegawa2010, Reference Hasegawa2016), 18S rDNA and cox1 gene sequences for identifying S. stercoralis in humans and dogs (Jaleta et al., Reference Jaleta2017), and cox1, 18S rDNA and 28S rDNA sequences for identifying S. stercoralis in humans and dogs (Nagayasu et al., Reference Nagayasu2017). Recently, S. fuelleborni has been reported from humans having contact with long-tailed macaques in Thailand (Thanchomnang et al., Reference Thanchomnang2017). Strongyloides fuelleborni has been identified, based on morphological criteria, in long-tailed macaques (M. fascicularis) from north-east Thailand (Wenz-Mücke et al., Reference Wenz-Mücke2013) and confirmed by molecular identification in the present study. These monkeys are now mostly found around temples or recreation parks near human communities (Malaivijitnond & Hamada, Reference Malaivijitnond and Hamada2008).
In conclusion, molecular evidence of S. fuelleborni infection in the long-tailed macaque in Thailand and Lao PDR has been presented. The common species infecting humans, S. stercoralis, is highly prevalent in human populations in these areas (Laymanivong et al., Reference Laymanivong2016; Prasongdee et al., Reference Prasongdee2017). Clearly, awareness needs to be raised of the zoonotic potential of S. fuelleborni. Molecular data for systematic, taxonomic and diagnostic studies in human populations at risk of S. stercoralis and S. fuelleborni infection are important for continuing epidemiological investigations. Such data will also inform prevention and control programmes to reduce animal-to-human transmission in this region. A monitoring programme should be organized, taking into account the role of reservoir hosts (i.e. monkeys) in the natural background of human strongyloidiasis caused by S. fuelleborni.
Acknowledgements
We would like to thank David Blair for valuable suggestions and assistance with the presentation of this paper through Khon Kaen University Publication Clinic.
Financial support
This study was supported by a TRF Senior Research Scholar Grant, Thailand Research Fund (T.T., P.M.I. and W.M., grant number RTA5880001); Scholarship under Doctoral Training Program from Graduate School Research Affairs and Khon Kaen University (C.S., grant number 59146) and the Faculty of Medicine, Mahasarakham University (T.T.).
Conflict of interest
None.
Ethical standards
All procedures contributing to this work comply with the ethical standards of the relevant national and institutional guides on the care and use of laboratory animals and were approved by the Khon Kaen University Ethics Committee for Animal Research (AEMDKKU 001/2018).