I. INTRODUCTION
Methylprednisolone acetate (marketed under the tradename Depo-Medrol®) is a synthetically manufactured corticosteroid medication (drug class corticosteroid hormone) administered primarily as an intramuscular, intra-articular, soft tissue, or intralesional injection in a dosage-dependent manner, or in pill form. It functions as an anti-inflammatory glucocorticoid, which is termed adrenocortical steroid. The use of analogous glucocorticoid pharmacology is a synthetic alternative often practiced in adrenocortical deficiency conditions. It is commonly used in the treatment of pain and swelling in arthritis patients (along with other joint disorders), certain cancers (leukemia, lymphoma, and multiple myeloma) alongside chemotherapy, as well as severe allergic reactions, and immune system and organ system disorders. Methylprednisolone acetate helps in reducing sickness during chemotherapy, aids in the reduction of immune system response, and is clinically proven to help the treatment of cancer itself. The IUPAC name (CAS Registry number 53-36-1) is [2-[(6S,8S,9S,10R,11S,13S,14S,17R)-1117-dihydroxy-6,1013-trimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-17-yl]-2-oxoethyl] acetate. A two-dimensional molecular diagram is shown in Figure 1.

Figure 1. The molecular structure of methylprednisolone acetate.
This work was carried out as part of a project (Kaduk et al., Reference Kaduk, Crowder, Zhong, Fawcett and Suchomel2014) to determine the crystal structures of large-volume commercial pharmaceuticals, and include high-quality powder diffraction data for these pharmaceuticals in the Powder Diffraction File (ICDD, 2017).
II. EXPERIMENTAL
Methylprednisolone acetate was a commercial reagent, purchased from USP (Lot # H0D148), and was analyzed as-received. The white powder was packed into a 1.5 mm diameter Kapton capillary, and rotated during the measurement at ~50 cycles s−1. The powder pattern was measured at 295 K at beam line 11-BM (Lee et al., Reference Lee, Shu, Ramanathan, Preissner, Wang, Beno, Von Dreele, Ribaud, Kurtz, Antao, Jiao and Toby2008; Wang et al., Reference Wang, Toby, Lee, Ribaud, Antao, Kurtz, Ramanathan, Von Dreele and Beno2008) of the Advanced Photon Source at Argonne National Laboratory using a wavelength of 0.413685 Å from 0.5–50° 2θ with a step size of 0.001° and a counting time of 0.1 s/step. The pattern was indexed on a primitive orthorhombic unit cell having a = 8.17523, b = 9.68097, c = 26.37087 Å, V = 2087.1 Å3, and Z = 4 using N-TREOR as incorporated in EXPO2014 (Altomare et al., Reference Altomare, Cuocci, Giacovazzo, Moliterni, Rizzi, Corriero and Falcicchio2013). Analysis of the systematic absences suggested the space group P212121, which was confirmed by successful solution and refinement of the structure. A reduced cell search in the Cambridge Structural Database (Groom et al., Reference Groom, Bruno, Lightfoot and Ward2016) combined with the chemistry “C H O only” yielded 26 hits, but no entry for methylprednisolone acetate.
The structure of the methylprednisolone acetate molecule was built and its conformation optimized using Spartan ’16 (Wavefunction, 2017). The resulting mol2 file was converted into a Fenske-Hall Z-matrix file using OpenBabel (O'Boyle et al., Reference O'Boyle, Banck, James, Morley, Vandermeersch and Hutchison2011). The structure was solved with FOX (Favre-Nicolin and Černý, Reference Favre-Nicolin and Černý2002). The maximum sinθ/λ used in the structure solution was 0.4 Å−1. A total of 59 cycles at two million trials/run were carried out. The lowest cost factor was the very low value 32 113. Many runs had similarly low cost factors.
Rietveld refinement was carried out using GSAS (Toby, Reference Toby2001; Larson and Von Dreele, Reference Larson and Von Dreele2004). Only the 1.7–25.0° portion of the pattern was included in the refinement (d min = 0.955 Å). All non-H bond distances and angles were subjected to restraints, based on a Mercury/Mogul Geometry Check (Sykes et al., Reference Sykes, McCabe, Allen, Battle, Bruno and Wood2011; Bruno et al., Reference Bruno, Cole, Kessler, Luo, Motherwell, Purkis, Smith, Taylor, Cooper, Harris and Orpen2004) of the molecule. The Mogul average and standard deviation for each quantity were used as the restraint parameters. The restraints contributed 1.9% to the final χ 2. The hydrogen atoms were included in calculated positions, which were recalculated during the refinement using Materials Studio (Dassault, 2016). A common U iso was refined for the non-H atoms of the ring system, another U iso for the non-H substituent atoms, and a third U iso for the acetate side chain. The U iso for each hydrogen atom was constrained to be 1.3 × that of the heavy atom to which it is attached. The peak profiles were described using profile function #4 (Thompson et al., Reference Thompson, Cox and Hastings1987; Finger et al., Reference Finger, Cox and Jephcoat1994), which includes the Stephens (Reference Stephens1999) anisotropic strain broadening model. The background was modeled using a three-term shifted Chebyshev polynomial, with a six-term diffuse scattering function to model the Kapton capillary and any amorphous component. The final refinement of 114 variables using 23 388 observations (23 303 data points and 85 restraints) yielded the residuals Rwp = 0.0840, Rp = 0.0668, and χ2 = 1.497. The largest peak (1.61 Å from C59) and hole (also 1.61 Å from C59) in the difference Fourier map were 0.61 and −0.61 eÅ−3, respectively. The Rietveld plot is included as Figure 2. The largest errors in the fit are in the shapes and positions of some of the low-angle peaks, perhaps reflecting specimen decomposition.

Figure 2. (Colour online) The Rietveld plot for methylprednisolone acetate. The black crosses represent the observed data points, and the red line is the calculated pattern. The blue curve is the difference pattern, plotted at the same vertical scale as the other patterns. The vertical scale has been multiplied by a factor of 10 for 2θ > 7.0°, and by a factor of 40 for 2θ > 14.3°.
A density functional geometry optimization (fixed experimental unit cell) was carried out using CRYSTAL09 (Dovesi et al., Reference Dovesi, Orlando, Civalleri, Roetti, Saunders and Zicovich-Wilson2005). The basis sets for the H, C, and O atoms were those of Gatti et al. (Reference Gatti, Saunders and Roetti1994). The calculation was run using 8 k-points and the B3LYP functional, and took ~12 days on a 2.8 GHz PC.
III. RESULTS AND DISCUSSION
The synchrotron powder pattern [Figure 3(a)] is similar enough to those of USP reference methylprednisolone acetate and commercial Depo-Medrol® suspension injection from Chinese Patent Application CN104710493A (He and Han, Reference He and Han2015) to conclude that they are the same material [Figure 3(b–c), digitized using UN-SCAN-IT 7.0 (Silk Scientific, 2013)]. All three patterns are similar enough to the pattern of form II in the same patent application to conclude that they all represent form II. The same form has also been obtained by recrystallization from tetrahydrofuran, acetone, and methanol by Sacha et al. (Reference Sacha, Schmitt and Nail2006) [Figure 3(d–e)].

Figure 3. (Colour online) Comparison of (a) the synchrotron pattern of methylprednisolone acetate from this study to those of (b) USP reference methylprednisolone and (c) commercial Depo-Medrol® suspension injection and to two patterns of form II (d), (e). Plots 3b–e from Chinese Patent Application CN104710493A.
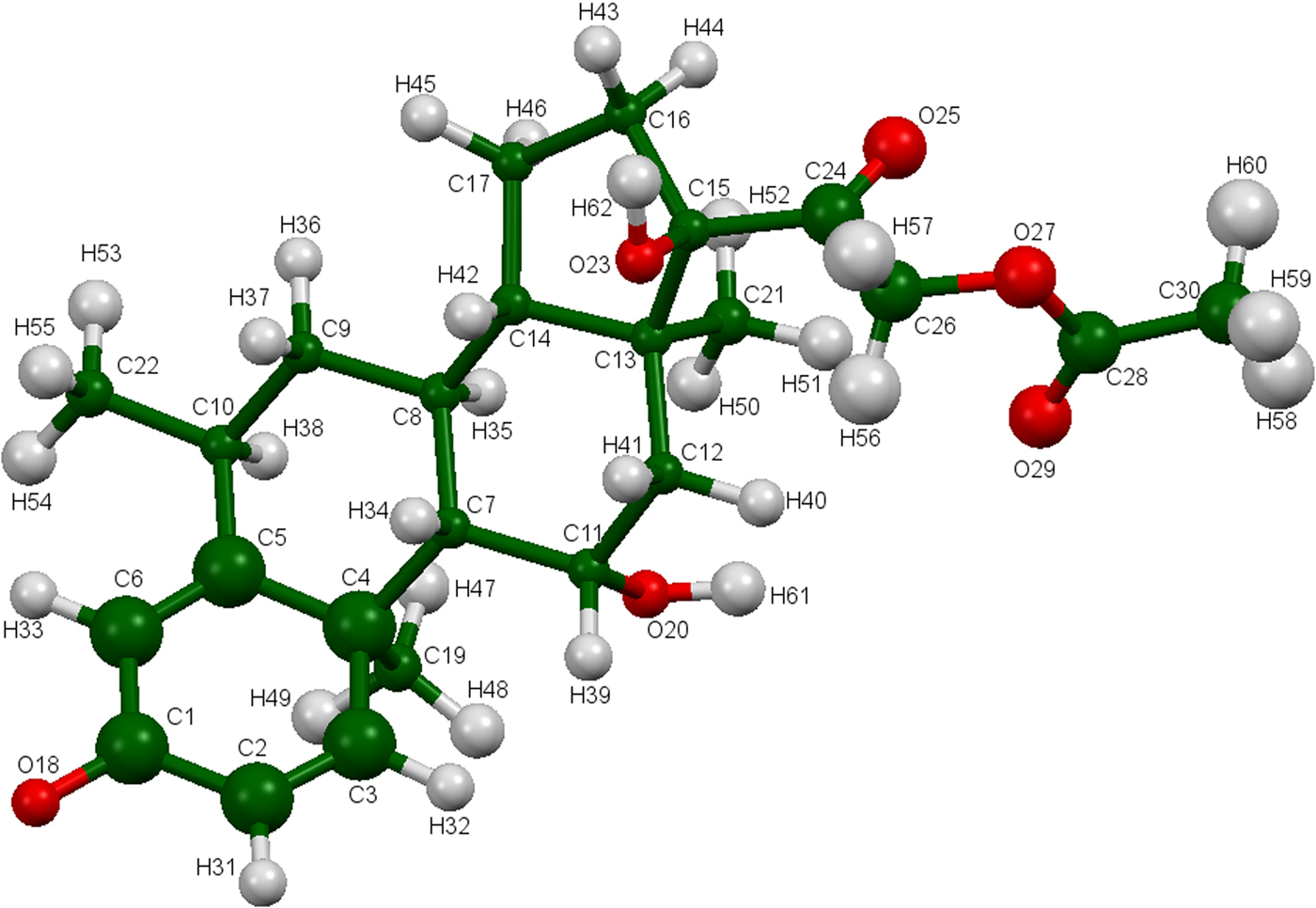
The refined atom coordinates of methylprednisolone acetate and the coordinates from the DFT optimization are reported in the Crystallographic Information Frameworks (CIFs) attached as Supplementary Material. The root-mean-square deviation of the non-hydrogen atoms in the methylprednisolone acetate molecules is 0.091 Å (Figure 4). The largest deviation is 0.234 Å at the hydroxyl group O20. The excellent agreement between the refined and optimized structures is evidence that the experimental structure is correct (van de Streek and Neumann, Reference van de Streek and Neumann2014). The following discussion uses the DFT-optimized structure. The asymmetric unit (with atom numbering) is illustrated in Figure 5, and the crystal structure is presented in Figure 6.

Figure 4. (Colour online) Comparison of the refined and optimized structures of methylprednisolone acetate. The Rietveld refined structure is in red, and the DFT-optimized structure is in blue.

Figure 5. (Colour online) The asymmetric unit of methylprednisolone acetate, with the atom numbering. The atoms are represented by 50% probability spheroids.

Figure 6. (Colour online) The crystal structure of methylprednisolone acetate, viewed down the a-axis.
All of the bond distances, bond angles, and torsion angles fall within the normal ranges indicated by a Mercury Mogul Geometry check (Macrae et al., Reference Macrae, Bruno, Chisholm, Edington, McCabe, Pidcock, Rodriguez-Monge, Taylor, van de Streek and Wood2008). Quantum chemical geometry optimizations (DFT/6-31G*/water) using Spartan ‘16 (Wavefunction, 2017) indicated that the observed conformation of methylprednisolone acetate is within 1 kcal/mole of the local minimum energy conformation. Molecular mechanics conformational analysis indicated that the global minimum energy conformation has a different orientation of the acetate group. The conformational analysis showed that several different orientations of the acetate had energies within 1 kcal/mole of the minimum energy conformation. Since the acetate does not participate in hydrogen bonds, we cannot rule out other orientations in the crystal structure but see no evidence for them.
Analysis of the contributions to the total crystal energy using the Forcite module of Materials Studio (Dassault, 2016) suggests that angle, bond, and torsion distortion terms are significant in the intramolecular deformation energy, as might be expected from a fused ring system. The intermolecular energy contains significant contributions from electrostatic attractions, which in this force-field-based analysis including hydrogen bonds. The hydrogen bonds are better analyzed using the results of the DFT calculation.
The hydroxyl group O23-H62 donates a proton to the hydroxyl group O20, to form a chain with graph set (Etter, Reference Etter1990; Bernstein et al., Reference Bernstein, Davis, Shimoni and Chang1995; Shields et al., Reference Shields, Raithby, Allen and Motherwell2000) C1,1(7) along the a-axis (Table I). The hydroxyl group O20-H61 donates a proton to the carbonyl oxygen atom O18 to form a chain with graph set C1,1(9) along the b-axis. The result is a two-dimensional hydrogen bond network in the ab plane. Three C–H⋯O hydrogen bonds (one intramolecular) also contribute to the crystal energy.
Table I. Hydrogen bonds in methylprednisolone acetate.

a Intramolecular.
The volume enclosed by the Hirshfeld surface (Figure 7; Hirshfeld, Reference Hirshfeld1977; McKinnon et al., Reference McKinnon, Spackman and Mitchell2004; Spackman and Jayatilaka, Reference Spackman and Jayatilaka2009; Wolff et al., Reference Wolff, Grimwood, McKinnon, Turner, Jayatilaka and Spackman2012) is 513.98 Å3, 98.6% of 1/4 the unit-cell volume. The molecules are thus not tightly packed. Some of the significant close contacts (red in Figure 7) involve the hydrogen bonds, while others indicate other close contacts.

Figure 7. (Colour online) The Hirshfeld surface of methylprednisolone acetate. Intermolecular contacts longer than the sums of the van der Waals radii are colored blue, and contacts shorter than the sums of the radii are colored red. Contacts equal to the sums of radii are white.
The Bravais–Friedel–Donnay–Harker (Bravais, Reference Bravais1866; Friedel, Reference Friedel1907; Donnay and Harker, Reference Donnay and Harker1937) morphology suggests that we might expect platy morphology for methylprednisolone acetate, with {002} as the principal faces. A second-order spherical harmonic preferred orientation model was included in the refinement; the texture index was 1.0004, indicating that preferred orientation was not significant in this rotated capillary specimen. The powder pattern of methylprednisolone acetate is included in the Powder Diffraction File as entry 00-065-1412.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715617001233
ACKNOWLEDGEMENTS
Use of the Advanced Photon Source at Argonne National Laboratory was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This work was partially supported by the International Centre for Diffraction Data. The authors thank Lynn Ribaud for his assistance in data collection.
FUNDING
Partial funding was provided by the International Centre for Diffraction Data, under Grant-in-Aid 09-03.
CONFLICT OF INTERESTS
The authors have no conflicts of interest to declare.