Introduction
Among cyclophyllidean cestodes, Hymenolepididae Perrier, 1897 is the most species-rich family comprising more than 920 species parasitic in birds and mammals (Mariaux et al., Reference Mariaux, Tkach, Vasileva, Caira and Jensen2017). The number of species from mammalian hosts exceeds 366 (Mariaux et al., Reference Mariaux, Tkach, Vasileva, Caira and Jensen2017; Makarikov et al., Reference Makarikov, Stakheev and Tkach2018, Reference Makarikov, Galbreath, Eckerlin and Hoberg2020; Makarikova, Reference Makarikova2018; Tkach et al., Reference Tkach, Kinsella and Greiman2018; Gardner et al., Reference Gardner, Dursahinhan, Campbell and Rácz2020; Makarikov & Georgiev, Reference Makarikov and Georgiev2020). These are parasitizing mostly insectivores (Eulipotyphla), rodents (Rodentia) and bats (Chiroptera) (Vaucher, Reference Vaucher1971; Czaplinski & Vaucher, Reference Czaplinski, Vaucher, Khalil, Jones and Bray1994; Georgiev et al., Reference Georgiev, Bray, Littlewood, Morand, Krasnov, Poulin, Morand, Krasnov and Poulin2006; Mariaux et al., Reference Mariaux, Tkach, Vasileva, Caira and Jensen2017). Phylogenetic relationships among hymenolepidids, including among the taxa occurring in mammals, remain unresolved. The pioneer study by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010) proposed the first phylogenetic hypothesis for the relationships among mammalian hymenolepidids, which was based on sequencing partial (D1–D3) 28S ribosomal RNA (rRNA) gene; it revealed the presence of four major phyletic lineages in the group, which were named ‘Ditestolepis clade’, ‘Arostrilepis clade’, ‘Hymenolepis clade’ and ‘Rodentolepis clade’. Subsequently, Neov et al. (Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019) analysed the phylogenetic relationships of this group based on partial (D1–D3) 28S rRNA gene of 12 selected taxa as well as sequences obtained by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010) and other authors (Greiman & Tkach, Reference Greiman and Tkach2012; Greiman et al., Reference Greiman, Tkach and Cook2013; Tkach et al., Reference Tkach, Makarikov and Kinsella2013, Reference Tkach, Kinsella and Greiman2018; Binkienė et al., Reference Binkienė, Kornienko and Tkach2015, Reference Binkienė, Miliūtė and Stunžėnas2019; Makarikov et al., Reference Makarikov, Mel'nikova and Tkach2015, Reference Makarikov, Stakheev and Tkach2018), comprising a total of 40 taxa. This study confirmed the same major clades but also added more details on the evolution of the host–parasite associations and the main morphological characteristics of the members of this group. Recently, using also 28S rRNA gene, Kornienko et al. (Reference Kornienko, Binkienė, Dokuchaev and Tkach2019) analysed the phylogenetic relationships within the Ditestolepis clade, involving seven out of the eight genera belonging to it. All these studies used homologous regions of the 28S rRNA gene. However, these results are only the beginning of understanding the evolutionary history of mammalian hymenolepidids. Generally, for more robust phylogenetic hypotheses of cestode groups, it is necessary to implement denser taxon sampling and the inclusion of additional genes (Mariaux & Olson, Reference Mariaux, Olson, Littlewood and Bray2001; Littlewood et al., Reference Littlewood, Waeschenbach and Nikolov2008; Waeschenbach & Littlewood, Reference Waeschenbach, Littlewood, Caira and Jensen2017; Kornienko et al., Reference Kornienko, Binkienė, Dokuchaev and Tkach2019).
The aim of the present study is to test the hypothesis for the phylogenetic relationships among mammalian hymenolepidids (based on the 28S rRNA gene) by examining the phylogeny of the group on the basis of cytochrome c oxidase I (COI) and 18S rRNA genes as well as a combined analysis using these two genes and the previously published sequences of 28S rRNA genes (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019).
Materials and methods
Cestode sampling and identification
The materials used in the present study were collected and analysed for 28S rRNA gene in a previous study (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019). Shrews were collected by trapping from Boyana River, Vitosha Mts. (42.637°, 23.260°) and Kalimok Field Station (44.012°, 26.440°) near Nova Cherna, Bulgaria. The helminthological study of host individuals was permitted by the Ministry of Environment and Waters of Bulgaria and followed the instructions presented in the permission. Adult cestodes were isolated from intestines. Specimens were preserved in 70% ethanol permitting both morphological and molecular study. Each cestode included in the analysis was divided into two parts. The anterior part (containing the scolex) was stained with iron acetocarmine (Georgiev et al., Reference Georgiev, Biserkov and Genov1986) and dehydrated in alcohol series, cleared in dimethyl phthalate and mounted in Canada balsam or Berlese's medium (Swan, Reference Swan1936) for morphological identification. Specimens used for DNA extraction were deposited as voucher slides in the Helminthological Collection of the Institute of Biodiversity and Ecosystem Research, Bulgarian Academy of Sciences (IBER–BAS), Sofia (table 1). The posterior parts of the specimens were used as tissue samples for DNA extraction.
Table 1. Cestode species sequenced and used in the phylogenetic analyses in the course of the present study.

a Accession numbers of the specimens used for DNA extraction (‘hologenophores’, see Pleijel et al., Reference Pleijel, Jondelius, Norlinder, Nygren, Oxelman, Schander, Sundberg and Thollesson2008) in the IBER–BAS Helminthological Collection are presented.
DNA extraction, polymerase chain reaction (PCR) amplification and sequencing
Total DNA was isolated using Single Worm PCR Protocol (Williams et al., Reference Williams, Schrank, Huynh, Shownkeen and Waterston1992). The amplification of a region of 18S rRNA gene was accomplished using the primers WormA 5′-GCGAATGGCTCATTAAATCAG-3′ (forward) and WormB 5′-CTTGTTACGACTTTTACTTCC-3′ (reverse) as suggested by Littlewood & Olson (Reference Littlewood, Olson, Littlewood and Bray2001). A section of the mitochondrial COI gene was amplified using the primers PBI-cox1F_PCR 5′-CATTTTGCTGCCGGTCARCAYATGTTYTGRTTTTTTGG-3′ (forward) and PBI-cox1R_PCR 5′-CCTTTGTCGATACTGCCAAARTAATGCATDGGRAA-3′ (reverse) (Scholz et al., Reference Scholz, de Chambrier, Kuchta, Littlewood and Waeschenbach2013). The PCR mixtures contained 25 μL of NZYTaq II 2× Green Master Mix (Cat. No. MB358; Nzytech, Lisbon, Portugal), 1 μM of each primer (FOR/REV), 10 ng template DNA and PCR-grade water to a total volume of 50 μL. PCR reactions for the 18S rRNA amplicon were carried out under the following conditions: initial denaturation at 94°C for 5 min, 30 cycles (denaturation at 94°C for 30 s; primer annealing at 50°C for 30 s; extension at 72°C for 120 s) and final extension at 72°C for 10 min. PCR reactions for amplification of the fragment of the COI gene were identical, with the difference that the extension phase was reduced to 30 s. PCR products were visualized on 1% agarose gel with GreenSafe staining (NZYTech, Lisbon, Portugal) under ultraviolet light. Fragment size was determined using GeneRulerTM 100 bp Ladder Plus (Fermentas, Thermo Scientific, Waltham, USA). All amplicons were purified using the PCR/DNA Clean-Up Purification Kit (EURx Sp. z o.o. Gdansk, Poland) and sequenced in both directions by a PlateSeq kit (Eurofins Genomics, Ebersberg, Germany) using the PCR primers (for 18S rRNA and COI genes) and two additional internal primers (for 18S rRNA gene): 1270F 5′-ACTTAAAGGAATTGACGG-3′ and 1270R 5′-CCGTCAATTCCTTTAAGT-3′.
Phylogenetic analyses
The newly obtained 12 18S rRNA sequences and 12 COI sequences (table 1) were manually edited and then aligned using MEGA software, version 7.0 (Kumar et al., Reference Kumar, Stecher and Tamura2016) and version X (Kumar et al., Reference Kumar, Stecher, Li, Knyaz and Tamura2018). An analysis using Basic Local Alignment Search Tool (BLAST analysis, see www.ncbi.nlm.nih.gov/BLAST) was applied for comparison and possible identification with sequences available in GenBank for the family Hymenolepididae. For phylogenetic analyses, we used published sequences of 18S rRNA gene (table 2) and COI gene (table 3) from several previous studies (Littlewood & Olson, Reference Littlewood, Olson, Littlewood and Bray2001; Olson et al., Reference Olson, Littlewood, Bray and Mariaux2001, Reference Olson, Yoder, Fajardo LG, Marty, van de Pas, Olivier and Relman2003; Littlewood et al., Reference Littlewood, Waeschenbach and Nikolov2008; Guo, Reference Guo2016; Nkouawa et al., Reference Nkouawa, Haukisalmi, Li, Nakao, Lavikainen, Chen, Henttonen and Ito2016; Zhao et al., Reference Zhao, Wang, Jia, Zhao, Hu, Yu and Liu2016; Pistone et al., Reference Pistone, Lindgren, Holmstad, Ellingsen, Kongshaug, Nilsen and Skorping2017; Dimitrova et al., Reference Dimitrova, Georgiev, Mariaux and Vasileva2019). GenBank sequences with less than 90% length coverage compared to our dataset were excluded. The analyses involved 19 sequences for 18S rRNA gene and 22 sequences for the COI gene. The combined analysed involved all taxa originally sequenced for the purposes of the present study (table 1) as well as three additional taxa (Dilepis undula, Hymenolepis diminuta and Rodentolepis microstoma). These were totally 15 taxa, for which sequences with sufficient coverage for the three genes were available – that is, 18S rRNA and COI (tables 2 and 3) and 28S rRNA (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019).
Table 2. Published sequences of 18S rRNA gene of dilepidid (Dilepis undula, outgroup) and hymenolepidid cestodes deposited in GenBank used in the present phylogenetic analysis.

a The generic allocation of this species is uncertain. Pistone et al. (Reference Pistone, Lindgren, Holmstad, Ellingsen, Kongshaug, Nilsen and Skorping2017) mentioned it as a member of Hymenolepis; this genus currently includes mammalian cestodes only (e.g. Binkienė et al., Reference Binkienė, Miliūtė and Stunžėnas2019) and the affiliation of ‘H. microps’ requires further studies. Therefore, we designate it as a member of ‘Hymenolepis (sensu lato)’.
b For morphological and biological characteristics of the laboratory strain (‘Nottingham strain’), see Cunningham & Olson (Reference Cunningham and Olson2010).
Table 3. Published sequences of COI of dilepidid (Dilepis undula, outgroup) and hymenolepidid cestodes deposited in GenBank used in the present phylogenetic analysis.

a The identification of this sample from the Korean Peninsula requires further confirmation by morphological and molecular studies, since the original description of Hymenolepis hibernia is from Apodemus sylvaticus from Northern Ireland (Montgomery et al., Reference Montgomery, Montgomery and Dunn1987).
b For morphological and biological characteristics of the laboratory strain (‘Nottingham strain’), see Cunningham & Olson (Reference Cunningham and Olson2010).
Phylogenetic analyses were performed using Bayesian inference with MrBayes, version 3.2.7 (Ronquist et al., Reference Ronquist, Teslenko and van der Mark2012). Prior to analysis, the best model of nucleotide substitution was selected using MrModeltest 2.4 (Nylander et al., Reference Nylander, Ronquist, Huelsenbeck, Nieves-Aldrey and Buckley2004); in all the three cases, this was the general time reversible model, with gamma-distributed estimate of site rate variation and a portion of invariant sites (GTR + G + I). The analyses were each run for 1.5 × 107 generations, two separate runs, each with four chains, discarding 33% (5 × 106) of resulting trees as burn-in. As outgroup for phylogenetic reconstruction analyses of genetic data for the three genes, we used sequences of D. undula (Schrank, 1788), a species of the family Dilepididae, believed to represent the most closely related family-group taxon, for which matching molecular data were available (Mariaux et al., Reference Mariaux, Tkach, Vasileva, Caira and Jensen2017).
For clade groups revealed by the present analysis, we used the names proposed by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010) and adopted in our previous article (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019); however, in the majority of cases, additional taxa were added based either on our new data or published sequences by other authors. Average standard deviation of split frequencies below 0.01 was observed at the end of each run and served as a proof of chains reaching convergence. Branches persisting in less than 60% of post burn-in samples were treated as polytomies. Nodal support was expressed as posterior probabilities. The rate variation among sites was modelled with a gamma distribution (shape parameter = 1). There were a total of 2098 positions in the final dataset for the 18S rRNA analysis, 558 positions for the COI analysis and 3802 positions for the combined analysis.
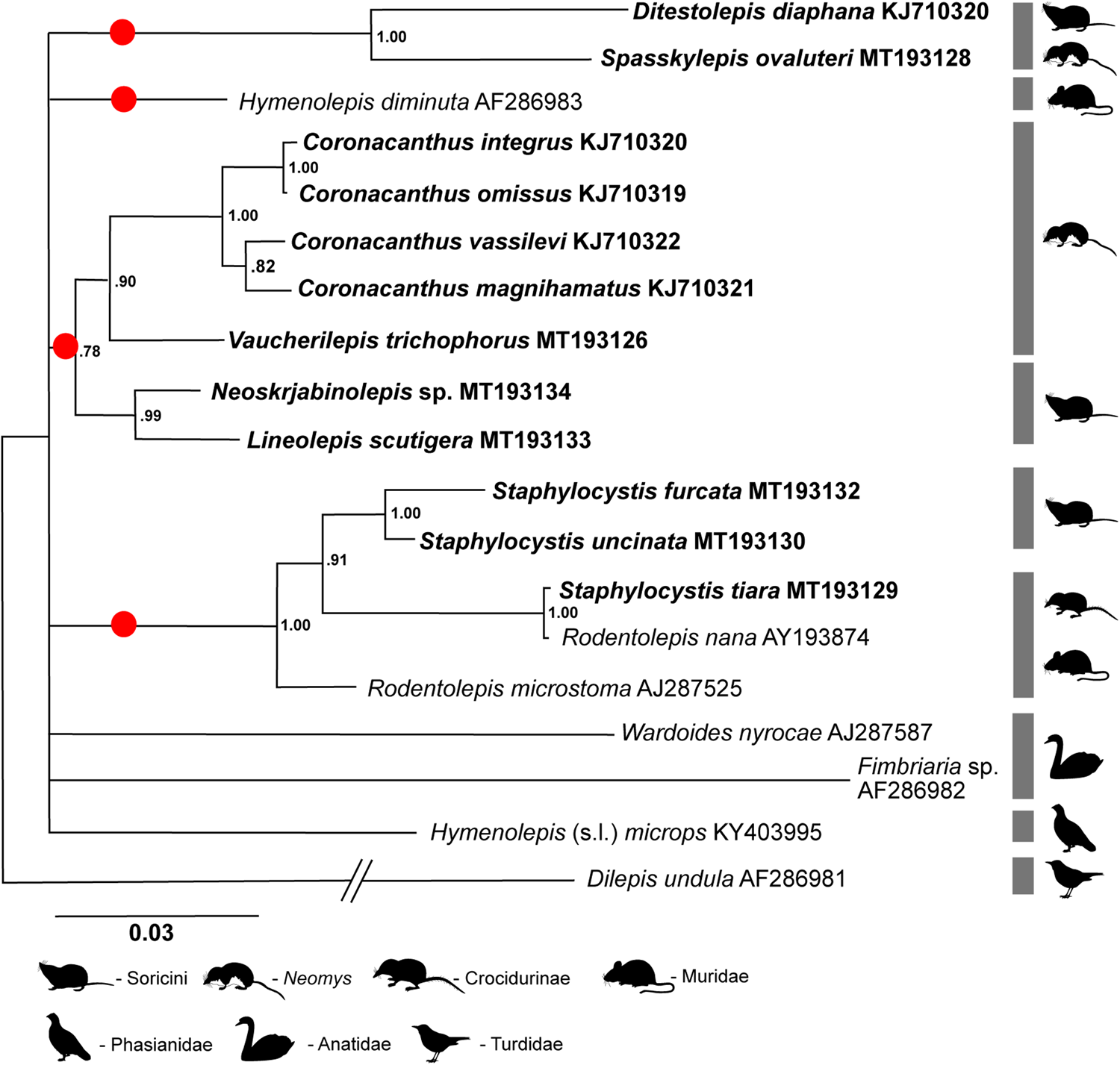
Results
Based on the 18S rRNA gene, basal relationships of the main phyletic lineages remained unresolved, with a polytomy of the main ‘mammalian’ clades forming with other hymenolepidids from birds, as well as with the only representative of the mammalian Hymenolepis clade (i.e. H. diminuta) included in the analysis (fig. 1). The two members of the Ditestolepis clade – that is, Ditestolepis diaphana and Spasskylepis ovaluteri – were revealed as sister taxa. The monophyly of the Arostrilepis clade was also well supported, with the genera Neoskrjabinolepis, Lineolepis, Vaucherilepis and Coronacanthus included in the present analysis. The monophyly of the genus Coronacanthus was also strongly supported, with another parasite of water shrews (Vaucherilepis) being its sister taxon. The Rodentolepis clade, represented by the genera Staphylocystis (parasitic in shrews) and Rodentolepis (parasitic in rodents) in the analysis, is also a monophyletic lineage, with R. microstoma basal to the remaining taxa and Rodentolepis nana (from rodents) and Staphylocystis tiara (from insectivores) revealed as sister taxa.

Fig. 1. Bayesian inference tree of phylogenetic relationships among 18 species of hymenolepidid cestodes (15 species from mammals and three species from birds) based on analysis of 18S rRNA gene. Dilepis undula (family: Dilepididae) is used as outgroup. The GenBank numbers are added after the binomial name of each species. Newly sequenced taxa are in bold. The major clades recognized by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010), confirmed by Neov et al. (Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019) and outlined by the present study are marked by circles. Nodal support is given by posterior probabilities. Scale bar shows the number of substitutions per site.
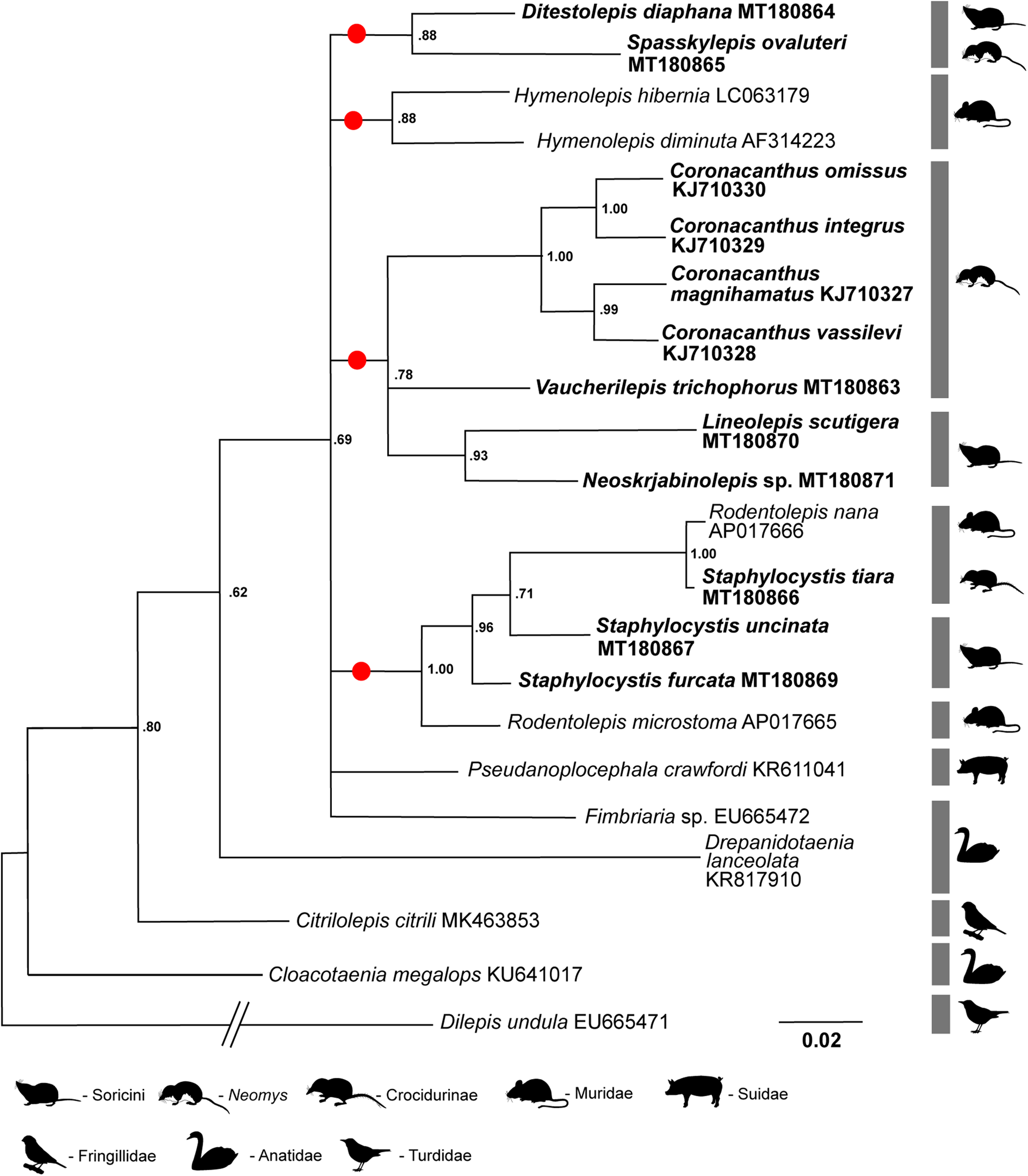
The COI analysis (fig. 2) revealed each of the four main clades of mammalian hymenolepidids as a monophyletic group. However, the basal branching of cestodes from small mammals remained unresolved, forming a polytomy with a hymenolepidid species parasitic in birds (Fimbriaria sp.) and the only hymenolepidid species parasitic in pigs (Pseudanoplocephala crawfordi). The remaining hymenolepidids from birds included in the analysis were basal to the polytomy that included taxa from mammals as well as Fimbriaria sp. The Ditestolepis clade and the Hymenolepis clade were well supported. The Arostrilepis clade was also well supported but the basal relationships in it were not resolved and the cestodes from water shrews did not form a monophyletic group; however, the genera Neoskrjabinolepis and Lineolepis were confirmed as sister taxa. In the Rodentolepis clade, R. microstoma was the basal taxon, and Staphylocystis furcata was basal to its congeners and R. nana. The strong phylogenetic relationship between R. nana (from rodents) and S. tiara (from insectivores), as revealed by the 18S rRNA gene analysis, was confirmed.

Fig. 2. Bayesian inference tree of phylogenetic relationships among 21 species of hymenolepidid cestodes (17 species from mammals and four species from birds) based on analysis of COI gene. Dilepis undula (family: Dilepididae) is used as outgroup. The GenBank numbers are added after the binomial name of each species. Newly sequenced taxa are in bold. The major clades recognized by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010), confirmed by Neov et al. (Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019) and outlined by the present study are marked by circles. Nodal support is given by posterior probabilities. Scale bar shows the number of substitutions per site.
The combined analysis based on sequences of 28S rRNA, 18S rRNA and COI genes (fig. 3) was characterized by the presence of a basal polytomy. However, the Ditestolepis clade, the Arostrilepis clade and the Rodentolepis clade were each strongly supported. The arrangement of the taxa in the Arostrilepis clade was identical with that revealed by the 18S rRNA gene analysis. In the Rodentolepis clade, S. tiara was basal to the remaining taxa, which included both Staphylocystis spp. and R. microstoma.

Fig. 3. Bayesian inference tree of phylogenetic relationships among 14 species of mammalian hymenolepidid cestodes based on combining sequences of COI, 18S rRNA and 28S rRNA genes. Dilepis undula (family: Dilepididae) is used as outgroup. Taxa represented partially by new sequences are in bold. The major clades recognized by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010), confirmed by Neov et al. (Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019) and outlined by the present study are marked by circles. Nodal support is given by posterior probabilities. Scale bar shows the number of substitutions per site.
Discussion
The phylogenies based on the 18S rRNA and COI genes confirm the main monophyletic groups among mammalian hymenolepidids revealed by sequencing of 28S rRNA genes (Haukisalmi et al., Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010; Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019). The presence of three of the major clades (Ditestolepis clade, Arostrilepis clade and Rodentolepis clade) is supported by all the analyses performed in the course of the present study. The Hymenolepis clade is also supported by the COI-based analysis; however, the 18S rRNA analysis and the combined analysis are not informative about this lineage, since it is represented by a single species only. Of the four major clades, only the Ditestolepis clade has a strong host–parasite association with shrews of the family Soricidae (Haukisalmi et al., Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010; Kornienko et al., Reference Kornienko, Binkienė, Dokuchaev and Tkach2019; Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019). As previously shown (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019), each of the remaining three clades includes mostly parasites from rodents and soricids, and sometimes from other mammalian orders. This distribution of host–parasite associations across the phylogenetic trees indicates multiple events of host switching in the course of the diversification of hymenolepidids in mammals (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019). More detailed discussions on the distributions of the various patterns of rostellar apparatus, host–parasite associations and lifecycle peculiarities across the clades confirmed by the present study have been presented by Neov et al. (Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019).
The relationships within the Ditestolepis clade and the Hymenolepis clade cannot be discussed confidently due to the limited number of taxa included in each. The relationships within the Arostrilepis clade, especially those based on 28S rRNA gene (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019) and 18S rRNA gene as well as the combined analysis, confirm the close relationships of cestodes from water shrews of the genus Neomys, represented in the present dataset by two morphologically dissimilar (Genov, Reference Genov1980; Tkach et al., Reference Tkach, Vasileva and Genov2003; Vasileva et al., Reference Vasileva, Tkach and Genov2005) genera – that is, Vaucherilepis and Coronacanthus. This might be a basis to speculate that armed hymenolepidids from water shrews of the genus Neomys are a monophyletic group. However, this hypothesis needs to be tested on the basis of more diverse sample of species.
The position of S. tiara, a parasite from crocidurine shrews, varies across the performed analyses, although always positioned in the Rodentolepis clade. Two of the analyses (28S rRNA and combined; fig. 3) place it in basal position but the 18S rRNA and COI analyses group it with R. nana as a sister taxon, both having a derived position (figs 1 and 2). The variable position of S. tiara revealed by the present study and the polyphyletic character of the genera Staphylocystis Villot, 1877 and Rodentolepis Spasskii, 1954 revealed by the 28S rRNA analysis (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019) suggest the need for further studies to resolve the phylogenetic relationships of these taxa and to revise the generic concepts in the group.
The monophyly of the hymenolepidids from ‘rodents and shrews’ was postulated by Haukisalmi et al. (Reference Haukisalmi, Hardman, Foronda, Feliu, Laakkonen, Niemimaa, Lehtonen and Henttonen2010) and adopted by Neov et al. (Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019). However, two of the present analyses (18S rRNA and COI) show that the basal relationships of the four mammalian clades are branching at the same polytomy with several hymenolepidids parasitic in birds (both terrestrial and aquatic). This questions the monophyly of the hymenolepidids from mammals and may indicate multiple events of colonizations of mammalian hosts. Therefore, the position and the evolutionary history of mammalian hymenolepidids require a more comprehensive consideration involving taxa of this family from both birds and mammals.
Though differing by the number of the taxa included, the phylogenetic trees based on 28S rRNA gene (Neov et al., Reference Neov, Vasileva, Radoslavov, Hristov, Littlewood and Georgiev2019), 18S rRNA gene and the combined tree have similar topologies. Although partial (D1–D3) large (28S) and complete small (18S) nuclear ribosomal genes form part of a tandem array (Hillis & Dixon, Reference Hillis and Dixon1991), together and in combination with partial COI, this combination of genes has been shown to demonstrate utility in resolving intra- and intergeneric relationships among cestode orders (e.g. Waeschenbach & Littlewood, Reference Waeschenbach, Littlewood, Caira and Jensen2017). However, in this study, in spite of adding additional molecular phylogenetic signal from genes known to evolve faster (COI) and slower (18S rRNA) than 28S rRNA D1–D3 (e.g. Machida & Knowlton, Reference Machida and Knowlton2012), no further resolution was added to the overall molecular phylogenetic hypothesis of hymenolepidids from mammals. Whilst denser, perhaps phylogenomic, gene sampling may be required to resolve the early branching patterns of hymenolepidid lineages, unresolved basal lineages and short internal branches are also considered signatures of rapid bursts of speciation and phenotypic evolution (Schluter, Reference Schluter2000). Undoubtedly, as the most species-rich clade of tapeworms, hymenolepidid cestodes have been highly successful in parasitizing birds and mammals, and it may be that their appearance was followed by rapid expansion and radiation through these host lineages.
Acknowledgements
We are grateful to the staff of the Kalimok Field Station of the Institute of Biodiversity and Ecosystem Research, Bulgarian Academy of Sciences, for facilitating field studies. Sampling was permitted by the Ministry of Environment and Waters of the Republic of Bulgaria, licences NSZP-153/11.05.2012 and NSZP-350/11.09.2014.
Financial support
This work was partly funded by a research project included in the collaborative programme of the Russian Foundation for Basic Research (grant number 19-54-18015) and the National Science Fund of Bulgaria (grant number KP-06-Russia-06).
Conflicts of interest
None.
Ethical standards
Methodology of the small mammal examination was approved by the Scientific Council of Institute of Biodiversity and Ecosystem Research, Bulgarian Academy of Sciences, Decision 7/16.10.2012.