Significant outcome
∙ Combinations of genetic variants significantly associated to bipolar disorder can be found in a large percentage of the patients, but being completely absent in control subjects.
Limitation
∙ When the number of combinations of genetic variants is very high, only a minor part of the combinations in a data set can be analysed due to methodological limitations.
Introduction
Bipolar disorder is a polygenic disorder involving a complex interplay of environmental factors and several susceptibility genes. Each susceptibility gene makes only a marginal contribution to the risk. Thus, the genetic basis for a polygenic disorder is one or more combinations of genetic variants. Although this concept is not new, a genetic variant combination clearly related to a polygenic disorder has not been described, largely because formerly very few genetic variants were known, but today a huge number of genetic variants have been identified, facilitating the search for combinations.
Combinations of genetic variants constituting the basis for polygenic disorders are assumed to not normally be found in healthy persons who are genetically unrelated to the patients. This assumption underlies a research strategy in which genetic variant combinations occurring exclusively in patients are analysed. A supplementary research strategy is to look for combinations of genetic variants in genes related to the biology of a disorder. For example, in a study of oral cancer, some combinations of genetic variants in genes related to DNA repair were found to be significantly associated with the disorder and were present in 55% of the patients but completely absent from controls (Reference Mellerup, Moeller, Mondal and Roychoudhury1). In addition, combinations of genetic variants related to neuroblastoma in single locus studies were found to be significantly associated with the disorder. These combinations were present in 9% of all patients and 13% of high risk neuroblastoma patients but completely absent from controls (Reference Capasso, Calabrese, Iolascon and Mellerup2). Some combinations of genetic variants in genes related to signal transduction in the brain have been found to be significantly associated with bipolar disorder, these combinations were present in 34% of the patients but completely absent from controls (Reference Koefoed, Andreassen and Bennike3,Reference Mellerup, Andreassen and Bennike4).
Of the 446 729 SNPs analysed in the data set from The Wellcome Trust Case Control Consortium (5) regarding bipolar disorder, 469 were also analysed in the previous study of genes related to signal transduction in the brain (Reference Koefoed, Andreassen and Bennike3). Aims of the present study were to analyse combinations of SNP genotypes from The Wellcome Trust Case Control Study of bipolar disorder (5,Reference Sklar, Smoller and Fan6).
Materials and methods
Patients, controls, and genotyping
The patients, the diagnostic criteria, the controls, and the selection of SNPs have been described in detail by Sklar et al. (Reference Sklar, Smoller and Fan6).
Combinations
Scanning data for combinations is a simple task in principle, but it may be difficult in practice because a data set may contain many billions of possible combinations. Even relatively powerful computers may be unable to perform such a task. Increased computer power, parallel computing by graphics processing units (Reference Bottolo, Chadeau-Hyam and Hastie7,Reference Sluga, Curk, Zupan and Lotric8), and cloud computing (Reference Guo, Meng, Yu and Pan9,Reference Dong, Xu and Fu10) can decrease the scanning time for combinations and were used in the present study.
Specialised software can be helpful in analysing genetic variant combinations. Algorithms and data mining tools have been developed for this purpose based on methods such as regression analysis, Bayesian statistics, and Boolean algebra (Reference Wei, Hemani and Haley11). The present study used array-based mathematical methods in which data are represented geometrically (Reference Mellerup, Andreassen and Bennike4,Reference Grelck and Scholz12), facilitating ultrafast parallel processing.
If the number of possible combinations in a study of genetic variants is too high to be analysed using the available technical tools, various methods can be applied to select smaller subgroups of combinations. Thus, in the present study, in which the theoretical number of combinations of five SNP genotypes is 446 729!/5!(446 729–5)!×35=3.6×1028, χ2 tests were used to analyse each of the 446 729 SNP genotypes with regards to the distribution between patients and controls. SNP genotypes with low p-values were selected, and each of these SNP genotypes was paired with each of the other SNP genotypes to form combinations of two SNP genotypes. The p-values were chosen to be so low that the number combinations obtained can be handled by the computers. From these combinations, those with low p-values regarding the distribution between patients and controls were selected and paired with each of all the other SNP genotypes to form combinations of three SNP genotypes. This procedure was then repeated to form combinations of four and five SNP genotypes successively (Reference Mellerup, Andreassen and Bennike4). Among the combinations of five SNP genotypes, those that occur exclusively in patients were selected.
Another selection from the 446 729 SNPs was 469 SNPs belonging to genes related to signal transduction in the brain (Reference Koefoed, Andreassen and Bennike3). The theoretical number of combinations of five SNP genotypes taken from 469 SNPs is 469!/5!(469−5)!×35=4.5×1013. It was possible to scan the data set brute force for combinations of five SNP genotypes. The combinations that occur exclusively in patients were selected.
Statistics
Permutation tests can be used to analyse many different genetic variant combinations selected from a data set (Reference Pesarin and Salmaso13). In the present study, permutation tests were used to evaluate the assumption that, among the combinations found exclusively in patients, combinations common to many patients are more likely to be significantly associated with bipolar disorder than combinations found in few patients. In a permutation test, the null hypothesis is that the observed data are exchangeable with respect to groups – in this case, the patients and controls. For this analysis, indices for patients and controls were removed, and from the total group of subjects two random groups of pseudo-patients and pseudo-controls were created with the same sizes as the original groups. This was repeated 1000 times, and the combinations found exclusively in pseudo-patients and that were common to many pseudo-patients were identified in each of the 1000 permutations. The null hypothesis is validated if the number of pseudo-patients having these combinations in their genome is the same or higher than in the original data set in more than 50 of 1000 permutations (p>0.05), suggesting that the combinations found exclusively in patients that were also common to many patients may be random findings.
In a polygenic disorder with pronounced genetic heterogeneity, the number of patients exhibiting the same genetic variant combination may be too small to confirm a statistically significant association between a combination and the disorder. In such cases, clusters or subgroups of combinations that are common among many patients can be selected based on various criteria. For example, among combinations common to many patients, those combinations containing a common SNP genotype could be selected to form a cluster. For another type of subgrouping, a χ2 test could be used to analyse the SNP genotype distribution between patients and controls with the aim of forming a cluster of combinations containing an SNP genotype with a low p-value. A third possible method is to select clusters in which each combination contains an SNP genotype relating to a particular biological function or pathway (Reference Hall, Verma, Wallace, Lucas and Berg14).
Results
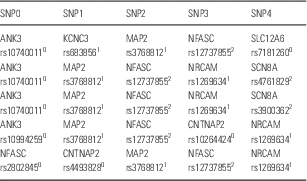
From the combinations of five SNP genotypes taken from the 469 SNPs related to signal transduction in the brain (Reference Koefoed, Andreassen and Bennike3), one cluster of combinations was found to be significantly associated with bipolar disorder (p<0.001). The cluster contained 68 combinations. Of the 1998 patients, 305 or 15% had some of these combinations in their genomes in contrast to none of the 1500 controls. Table 1 shows the first five combinations in the cluster. All 68 combinations and the 305 patients are shown in the Supplementary Table 1.
Table 1 The first five combinations of the cluster containing 68 combinations of five SNP genotypes
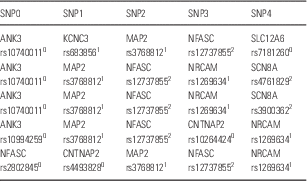
All 68 combinations are shown in Supplementary Table 1, which also shows the 305 patients who have some of these combinations in their genome. For each SNP, the rs number is given along with the protein: 0homozygous for the major allele, 1heterozygous, 2homozygous for the minor allele.
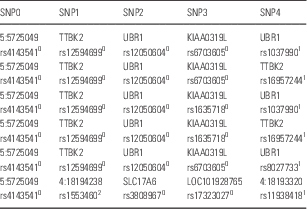
From the selected group of combinations of five SNP genotypes taken from the 446 729 SNPs, those that occurred exclusively in patients were extracted. A cluster containing six combinations was significantly associated with bipolar disorder (p<0.001). Of the 1998 patients, 515 (26%) had some of these combinations in their genomes, whereas none of the 1500 controls had any of the combinations in their genomes. Table 2 shows the cluster. The cluster and the 515 patients are shown in the Supplementary Table 2.
Table 2 Cluster containing six combinations of five SNP genotypes
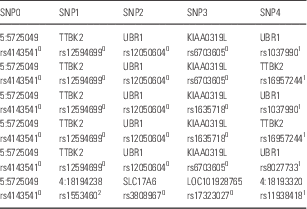
For each SNP, the rs number is given along with the protein or chromosomal location. TTBK2 is a tubulin kinase, UBR1 is a ubiquitin ligase, KIAA0319L is a receptor for adeno virus, and SLC17A6 is a glutamate transporter. 0homozygous for the major allele, 1heterozygous, 2homozygous for the minor allele. Supplementary Table 2 shows the rs numbers and dummy id for 515 patients who have some of these combinations in their genome.
No overlap of SNP genotypes was observed between the two clusters (Supplementary Tables 1 and 2), but 76 patients had combinations from both clusters in their genomes.
Discussion
Combinations of genetic variants are assumed to be the basis for polygenic disorders, and if such a combination is found it may be present in all patients suffering from the disorder, which corresponds to genetic homogeneity. In the case of genetic heterogeneity, various combinations may be the basis for a disorder in various patient subgroups. Due to the large number of possible combinations, an extreme degree of genetic heterogeneity in which every patient has a personal combination of genetic variants as a basis for the disorder cannot be excluded.
Supplementary Table 1 shows a cluster containing 68 combinations and 305 patients and each of these patients has some of the 68 combinations in the genome, but the combinations as well as the number of combinations may vary from patient to patient. Thus each of the 305 patients may have a personal pattern of combinations from the cluster in their genome (these patterns can be extracted from Supplementary Table 1). In the previous studies of bipolar patients from a Scandinavian sample in which combinations of three and four SNP genotypes were analysed, it was also found that each patient had a personal pattern of combinations in their genome (Reference Koefoed, Andreassen and Bennike3,Reference Mellerup, Andreassen and Bennike4). These personal patterns indicates an extreme degree of genetic heterogeneity in the bipolar patients. However, as can be seen from Supplementary Table 1 these patterns are very similar, hereby restoring a kind of genetic homogeneity. The same was found in the Scandinavian sample where the patterns of combinations were very similar within a single cluster of combinations, but showed differences between clusters (Reference Koefoed, Andreassen and Bennike3,Reference Mellerup, Andreassen and Bennike4). Relatively many of the patients contain these combinations, 15% of the Wellcome Trust sample and 34% of the Scandinavian sample, in contrast to 0% of the controls.
It is an often proposed hypothesis that changes in signal transduction in the brain may be related to bipolar disorder (Reference Koefoed, Andreassen and Bennike3), the personal patterns of combinations in the genomes of the patients suggest that a broad variety of changes in ion channels and other proteins involved in signal transduction may be related to bipolar disorder. It is interesting questions whether particular combinations leads to greater risk of bipolar disorder than other, or whether accumulation of many combinations in a genome leads to greater risk than accumulation of few combinations?
The clinical implication of these findings is that the SNPs found in the clusters can be analysed in a single patient, and the resulting SNP genotypes can be used to map the personal pattern of combinations in that patient. If some of these combinations are present in clusters associated to bipolar disorder it may support the diagnostic evaluation. Furthermore, patients belonging to a cluster can be seen as a genetic subgroup and it has been found that patients belonging to a particular cluster showed significantly more manic and depressive episodes than the patients belonging to other clusters (Reference Mellerup, Andreassen and Bennike15). Thus the personal pattern of combinations of SNP genotypes may indicate a possible clinical subtype of a patient.
The number of combinations of five SNP genotypes taken from all the 446 729 SNPs in the Wellcome Trust data set is too high (3.6×1028) to be analysed by brute force. Instead, combinations of SNP genotypes that occurred more often in patients than controls were analysed. This selection entails that the majority of combinations will remain unanalysed, and it cannot be excluded some of these combinations may be associated with bipolar disorder.
One cluster of six combinations of three SNP genotypes was found to be significantly associated with bipolar disorder. It is noteworthy that 26% of the patients had some of these combinations in their genomes, in contrast to none of the 1500 controls. This result is in line with the result from the cluster shown in Table 1, where 15% of the patients had combinations from the cluster in their genomes, and from a previous study of bipolar disorder (Reference Mellerup, Andreassen and Bennike4) where 34% of the patients had combinations from clusters in their genomes, and from a study of oral cancer (Reference Mellerup, Moeller, Mondal and Roychoudhury1) where 54% of the patients had combinations from clusters in their genomes.
The 446 729 SNPs in the data set were not selected on a basis related to bipolar disorder (Reference Sklar, Smoller and Fan6), because the SNPs were also analysed in many other types of patients (5), and a search in Medline and other databases did not reveal any association between the SNPs in the six combinations in Table 2 and bipolar disorder. However, this does not exclude an association, but the combinations may be a kind of genetic markers, and may not be directly involved in the biology of bipolar disorder.
The stepwise selection of combinations based on statistics left most of the combinations in the material unexamined, so selections based on other criteria may reveal more clusters of combinations significantly associated with bipolar disorder. Thus selections based on other biological criteria than signal transduction in the brain may be interesting to study.
In conclusion, studies of combinations of genetic variants can reveal combinations occurring exclusively in patients that are significantly associated with disorders, but it seems that so far only genetic variants related to the biology of a disorder may result in clinically useful combinations.
Acknowledgement
This study was supported by Beckett-Fonden, Copenhagen, Denmark.
Supplementary material
To view supplementary materials for this article, please visit https://doi.org/10.1017/neu.2017.36