


INTRODUCTION
Human African trypanosomiasis (HAT) is caused by the infection of Trypanosoma brucei (T. brucei) gambiense and T. brucei rhodesiense, which are transmitted to the human host by bites of tsetse flies. HAT threatens more than 70 million people, who live in endemic areas with 50 000 and 70 000 new infections per year (Welburn and Odiit, Reference Welburn and Odiit2002; World Health Organization, 2007) and is fatal if untreated. It is endemic mainly in sub-Saharan Africa, where the disease shows the highest prevalence rate, 60% in some villages in the Democratic Republic of the Congo (Smith et al. Reference Smith, Pepin and Stich1998; Welburn and Odiit, Reference Welburn and Odiit2002). HAT has been considered an essentially rural disease until recently. However, it has become a significant public health threat in large cities such as Brazzaville in the Congo Republic, Kinshasa in the Democratic Republic of the Congo and Conakry in the Guinea Republic (Smith et al. Reference Smith, Pepin and Stich1998; Bilengue et al. Reference Bilengue, Meso, Louis and Lucas2001). Therapy for HAT relies still upon drugs, i.e. pentamidine, suramin, melarsoprol (trivalent arsenical) and eflornithine (difluoromethyl-ornithine) that were first introduced over 50 years ago (Gutteridge, Reference Gutteridge1985; Doua and Yapo, Reference Doua and Yapo1993; Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007). Pentamidine and suramin are used for the treatment of the early haemolymphatic stages of HAT, whereas melarsoprol is used for treatment of the late central nervous system stages of the disease. However, the latter requires parenteral administration and causes a reactive encephalopathy in up to 10% of treated patients, with a mortality rate of up to 5%. Recently, 25–30% of patients in Central Africa with late-stage HAT have not responded to melarsoprol due most likely to the emergence of drug-resistant strains of the parasite (Ross and Sutherland, Reference Ross, Sutherland, Hide, Mottram, Coombs and Holmes1997). Eflornithine is effective for late-stage infections caused by T. brucei gambiense (Burri and Brun, Reference Burri and Brun2003), including many cases of arsenical resistance but ineffective against infections caused by T. brucei rhodesiense (Pepin and Milord, Reference Pepin and Milord1994; Iten et al. Reference Iten, Mett, Evans, Enyaru, Brun and Kaminsky1997; Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007). However, it requires the parenteral administration of large doses and also shows some toxic side-effects. Currently, there are no other drug candidates in the pharmaceutical pipelines for HAT. This uncertainty for a successful chemotherapy against HAT and the lack of immediate vaccine candidates against T. brucei infections add impetus to explore the basic metabolic pathways of trypanosomes in order to identify novel drug targets; the rational approach for a new drug design being the identification and exploration of targets which are unique and/or different from those of the host in the basic biochemistry of the parasites. One such biochemical pathway is the biosynthesis of guanine nucleotide that is a rich source of therapeutic targets. However, since many of the major metabolic pathways in eukaryotic parasites are similar or identical to those in their vertebrate host, it is generally difficult to devise selective therapies against these parasites. One metabolic pathway that is conspicuously different between parasites and humans is that for the production of purine nucleotides. Whereas mammalian cells synthesize purine nucleotides de novo, T. brucei is auxotrophic for purines and critically dependent on salvage mechanisms to acquire purine nucleotides from the extracellular environment.
Guanine nucleotides are involved in many cellular functions including transmembrane and intracellular signalling (Parandoosh et al. Reference Parandoosh, Robins, Belei and Rubalcava1989, Reference Parandoosh, Rubalcava, Matsumoto, Jolley and Robins1990; Kharbanda et al. Reference Kharbanda, Sherman and Kufe1990; Mandanas et al. Reference Mandanas, Leibowitz, Gharehbaghi, Tauchi, Burgess, Miyazawa, Jayaram and Boswell1993; Manzoli et al. Reference Manzoli, Billi, Gilmour, Martelli, Matteucci, Rubbini, Weber and Cocco1995), DNA replication, and RNA and protein synthesis (Jayaram et al. Reference Jayaram, Dion, Glazer, Johns, Robins, Srivastava and Cooney1982). De novo, guanosine 5′-monophosphate (GMP) is synthesized from inosine 5′-monophosphate (IMP). In this pathway, IMPDH (EC 1.1.1.205) catalyses the NAD-dependent oxidation reaction of IMP that is converted into xanthosine 5′-monophosphate (XMP) as shown in BOX 1 of Fig. 1. Subsequently, XMP is converted into GMP (BOX 2 of Fig. 1) by GMP synthase (EC 6.3.4.1 or 6.3.5.2). IMPDH is the rate-limiting enzyme in guanine nucleotide biosynthesis and also a key enzyme in the regulation of cell proliferation and differentiation (Wang and Hedstrom, Reference Wang and Hedstrom1997; Markham et al. Reference Markham, Bock and Schalk-Hihi1999; Digits and Hedstrom, Reference Digits and Hedstrom1999a). It has been the target of antiviral (Witkowski et al. Reference Witkowski, Robins, Sidwell and Simon1972; Patterson and Fernandez-Larsson, Reference Patterson and Fernandez-Larsson1990; Poynard et al. Reference Poynard, Marcellin, Lee, Niederau, Minuk, Ideo, Bain, Heathcote, Zeuzem, Trepo and Albrecht1998; Crotty et al. Reference Crotty, Maag, Arnold, Zhong, Lau, Hong, Andino and Cameron2000; Maag et al. Reference Maag, Castro, Hong and Cameron2001), antimicrobial, anti-cancer (Weber et al. Reference Weber, Prajda, Abonyi, Look and Tricot1996) and immunosuppressive (Wu, Reference Wu1994) therapies. IMPDHs have been found in almost every organism. In human, 2 isoenzymes of IMPDH have been identified, i.e. IMPDH type 1 which is constitutively expressed and IMPDH type 2 whose expression is up-regulated with peak enzyme activity occurring during the S phase of the cell cycle (Szekeres et al. Reference Szekeres, Fritzer, Pillwein, Felzmann and Chiba1992). All IMPDHs described so far are tetrameric proteins (Table S1, online version only). Literature and database search of trypanosome genome for the presence of IMPDH gene led to find a T. brucei gambiense nucleotide sequence of IMPDH gene (NCBI data bank ID: XM_822988) amplified due to an increase in chromosome copy number following treatment of this parasite with an excess amount of mycophenolic acid (MPA), an inhibitor of IMPDH (Wilson et al. Reference Wilson, Berens, Sifri and Ullman1994).
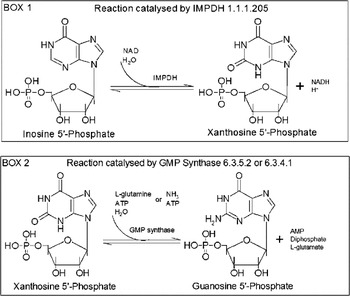
Fig. 1. Guanosine 5′-monophosphate biosynthetic pathway.
In this paper, we cloned and heterologously expressed TbIMPDH in Escherichia coli (E. coli), and showed that TbIMPDH is a unique large heptameric protein different from type 1 and 2 mammalian tetrameric IMPDHs. Kinetic analysis of TbIMPDH confirmed that the trypanosomal enzyme is indeed different from their mammalian counterparts, indicating that it is a good target for further studies on anti-trypanosomal drugs.
MATERIALS AND METHODS
Materials
IMP, NAD and MPA were purchased from Sigma Chemical Co. (St Louis, MO, USA). Ribavirin 5′-monophosphate (RMP) was purchased from Toronto Research Chemicals Inc. (North York, Canada). Mizoribine 5′-monophosphate (MZP) was synthesized as previously described (Shuto et al. Reference Shuto, Haramuishi, Fukuoka and Matsuda2000). PIC-A was purchased from Waters Co. (Milford, MA, USA) and diluted according to the manufacturer's manual to give a 5 mm tetrabutylammonium phosphate solution of pH 7·0. All other chemicals were of analytical grade.
Expression and purification of TbIMPDH
The full-length TbIMPDH gene (NCBI data bank ID: XM_822988) was PCR amplified from T. brucei Tb427 strain genomic DNA and ligated into the EcoRI-SalI sites of the expression vector pGEX-6P-1 plasmid (GE Healthcare BioScience, NJ, USA). Inserts of all clones were sequenced to reconfirm their sequences at the J Craig Venter Institute (JCVI, Rockville, MD, USA). E. coli BL21 (DE3) was transformed with the plasmid (TOYOBO, Tokyo, Japan) and the recombinant TbIMPDH was expressed as a GST fusion protein. The fusion protein was bound to glutathione-Sepharose 4B (GE Healthcare BioScience) and incubated with PreScission protease (GE Healthcare BioScience) to release TbIMPDH. The recombinant protein was further purified with a DEAE-TOYOPEARL anion exchange column (TOSOH, Tokyo, Japan). Protein purity was analysed by SDS-PAGE.
Size-exclusion chromatography
Size-exclusion chromatography was performed using the ÄKTA purifier HPLC apparatus equipped with a HiLoad 16/60 Superdex 200 pg gel-filtration column (GE Healthcare BioScience). The column was equilibrated and eluted with 50 mm Tris/Cl (pH 8·0) containing 150 mm KCl at a flow rate of 0·5 ml min−1. The column was calibrated with a mixture of thyroglobulin, ferritin, human IgG, human transferrin and ovalbumin (molecular masses are 669, 440, 150, 81 and 43 kDa, respectively). The protein elution was monitored by UV absorption at 280 nm.
AUC-SV experiment
AUC-SV experiments were performed in 50 mm potassium phosphate (pH 7·4) containing 150 mm KCl using a ProteomeLab XL-I Analytical Ultracentrifuge (Beckman-Coulter, Fullerton, CA, USA). Samples of TbIMPDH of different concentrations (4·8, 9·6 and 30 μ m) were measured. Centrifugations were carried out at 29, 46 and 66 × 103g at a temperature of 20 °C using a 12 mm filled-epon double sector centrepiece and four-hole An60 Ti analytical rotor equilibrated to 20 °C. Sedimentation was monitored with absorbance detection optics at appropriate wavelengths (226, 227 and 230 nm) according to the concentration of the solution and within the solution absorbance of between 0·8 and 1·3. The radial increment was 0·003 cm and at least 150 scans were collected between 5·9 and 7·25 cm from the centre of rotation axis. All SV raw data were analysed using a continuous C(s) distribution model with the SEDFIT11·71 program software (Schuck, Reference Schuck2000). The position of the meniscus and the frictional ratio (f/f 0) were set to vary as fitted parameters. A resolution of 300 increments between 0·1 and 50 S was entered and maximum entropy regularization was used (p = 0·68).
HPLC analysis of TbIMPDH products
The reaction mixture contained 50 mm Tris/Cl (pH 8·0), 100 mm KCl, 3 mm EDTA, 250 μ m IMP, 1 mm NAD and 150 nm enzyme. After filtering 500 μL of the reaction mixture using VIVASPIN 500 (Sartorius K.K, Tokyo, Japan), 10 μL of the filtrate were injected for HPLC analysis. HPLC analysis was carried out using an Agilent 1100 series HPLC system (Agilent Technologies, Waldbronn, Germany) equipped with a quaternary pump, an online degasser, a column oven, and a diode-array detector. A Cosmosil 5C18 MS-II column (2·0 mm × 150 mm, 5 μm, Nacalai Tesque, Kyoto, Japan) was used for separation at a column temperature of 35 °C. The mobile phase consisted of acetonitrile containing 0·1% trifluoroacetic acid (solvent A) and water containing 5 mm PIC-A reagent (solvent B). The gradient programmer was used according to the following procedure: 0–5 min hold on 5% A, 5–15 min linearly increase A from 5 to 50%, 15–20 min hold on 95% A, 20–30 min hold on 5% A with a flow-rate of 300 μL min−1. The UV detection wavelength was set at 254 nm.
Dependence of TbIMPDH activity on pH
Measurements of TbIMPDH activity at different pH were performed in the following buffers: sodium acetate (pH 4·0–6·0), sodium phosphate (pH 6·0–7·5), Tris/Cl (pH 7·5–9·0), CHES (pH 9·0) and carbonate (pH 10·0). The ionic strengths of these buffers were adjusted to 0·25 by adding NaCl. Reactions were initiated by adding IMP in a reaction mixture of 500 μ m IMP, 1 mm NAD and 150 nm enzyme and incubating at 25 °C for 10–40 min.
Enzyme kinetics
Standard IMPDH buffer contained 50 mm Tris/Cl (pH 8·0), 100 mm KCl, 3 mm EDTA and 1 mm DTT. The concentrations of IMP and NAD were varied to determine kinetic parameters. Reactions were initiated by the addition of IMP after the pre-incubation of the reaction mixture at 25 °C for 5 min and the activity was assayed in the presence of 200 nM enzyme at 25 °C for 10–40 min. NADH production was monitored at 340 nm (ε 340 = 6220 m−1 cm−1) and 25 °C using a UV-1600PC spectrophotometer (Shimadzu, Kyoto, Japan). Since substrate inhibition was observed, the initial velocity data were fitted into the Michaelis–Menten equation and uncompetitive substrate inhibition Equation 1 using Origin 6·0 software (Microcal, Northampton, MA, USA).
 $$v = V_{\rm m} /(1{\rm +} K_{\rm m} /[{\rm S}] + [{\rm S}]/K_{{\rm ii}} )$$
$$v = V_{\rm m} /(1{\rm +} K_{\rm m} /[{\rm S}] + [{\rm S}]/K_{{\rm ii}} )$$where: K m and K ii represent the Michaelis constant and substrate inhibition constant, respectively.
Inhibitor kinetics
For the K i value for RMP, experiments were carried out in the presence of constant concentrations of NAD (1·5 mm) but varied concentrations of RMP and IMP. For the K i value for MPA, reactions were performed in the presence of 300 μ m IMP and varied concentrations of MPA and NAD. The K i value for MZP was measured in reaction mixtures containing 160 μ m IMP and 1·5 mm NAD, and varied MZP concentrations. All the reaction mixtures contained 200 nm enzyme and were incubated at 25 °C for 5 min.
Data analysis for inhibition studies
Initial rate data were fitted to the competitive inhibition Equation 2, uncompetitive inhibition Equation 3 and the slow tight-binding competitive inhibition Equations 4 and 5 (Kerr and Hedstrom, Reference Kerr and Hedstrom1997) using Origin 6·0 software. The best fits were determined by the relative fit error.
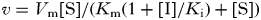 $$v = V_{\rm m} [{\rm S}]/(K_{\rm m} (1 + [{\rm I}]/K_{\rm i} ) + [{\rm S}])$$
$$v = V_{\rm m} [{\rm S}]/(K_{\rm m} (1 + [{\rm I}]/K_{\rm i} ) + [{\rm S}])$$ $$v = V_{\rm m} [{\rm S}]/(K_{\rm m} + [{\rm S}](1 + [{\rm I}]/K_{\rm i} ))$$
$$v = V_{\rm m} [{\rm S}]/(K_{\rm m} + [{\rm S}](1 + [{\rm I}]/K_{\rm i} ))$$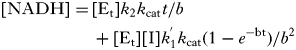 $$\eqalign{[{\rm NADH}] = \tab [{\rm E}_{\rm t} ]k_2 k_{{\rm cat}} t/b \cr \tab+ [{\rm E}_{\rm t} ][{\rm I}]k^' _1 k_{{\rm cat}} (1 - e^{ - {\rm bt}} )/b^2 $$}
$$\eqalign{[{\rm NADH}] = \tab [{\rm E}_{\rm t} ]k_2 k_{{\rm cat}} t/b \cr \tab+ [{\rm E}_{\rm t} ][{\rm I}]k^' _1 k_{{\rm cat}} (1 - e^{ - {\rm bt}} )/b^2 $$} $$b = [{\rm I}]k^' _1 + k_2 $$
$$b = [{\rm I}]k^' _1 + k_2 $$[Et] is the total concentration of enzyme, and [I] and K i are the total concentration of inhibitor and the inhibition constant, respectively. k 1′ is the observed association rate constant for inhibitor binding to enzyme, k 2 is the dissociation rate of the inhibitor enzyme complex, and k cat is the observed catalytic rate constant. The true association rate constant for inhibitor binding to the enzyme was obtained from Equation 6 and K i was calculated with Equation 7:
 $$k_1 = (K_{\rm m} + [{\rm IMP}])k^' _1 /K_{\rm m} $$
$$k_1 = (K_{\rm m} + [{\rm IMP}])k^' _1 /K_{\rm m} $$ $$K_{\rm i} = k_2 /k_1 $$
$$K_{\rm i} = k_2 /k_1 $$RESULTS
Characterization of TbIMPDH subunit structure
We searched the literature and database of the trypanosome genome for the presence of IMPDH gene and found a T. b. gambiense nucleotide sequence of IMPDH gene (NCBI data bank ID: XM_822988) that had been amplified due to an increase in chromosome copy number following treatment of T. b. gambiense with an excess amount of MPA (Wilson et al. Reference Wilson, Berens, Sifri and Ullman1994); an inhibitor of IMPDH. We cloned and heterologously expressed this IMPDH gene. The purified recombinant TbIMPDH yielded a discrete single homogenous protein band on SDS-PAGE. The monomeric molecular mass of TbIMPDH was found to be 57 kDa (Fig. 2). Size-exclusion chromatography analysis revealed that the estimated molecular mass of the native TbIMPDH was approximately 420 kDa (Fig. 3A).

Fig. 2. SDS-PAGE analysis of purified TbIMPDH. The purified TbIMPDH of 3 μg was analysed by SDS-PAGE. The migration positions of the molecular mass standards are indicated on the right side. Marker proteins myosin (200 kDa), β-galactosidase (116 kDa), albumin (66 kDa), ovalbumin (45 kDa) and carbonic anhydrase (31 kDa) were used. Bands were visualized by staining the gel with Coomassie brilliant blue.

Fig. 3. Subunit structure of TbIMPDH. (A) Size-exclusion chromatography analysis of purified TbIMPDH. TbIMPDH was loaded onto a HiLoad 16/60 Superdex 200 pg gel-filtration column and eluted at the elution volume of 56·6 mL. Inset shows the calibration curve of the HiLoad 16/60 Superdex 200 pg gel filtration. Molecular mass standards: thyroglobulin (filled circle), ferritin (filled square), human IgG (open square), human transferrin (filled triangle) and ovalbumin (open triangle) were used. (B) Distribution states of TbIMPDH analysed by AUC-SV. Results of C(s) analysis at different TbIMPDH concentrations are shown. The sedimentation coefficient was converted into s 20,w.
Since the quaternary structure of the trypanosome enzyme was unusually large, we investigated the assembly state of TbIMPDH using analytical ultracentrifugation and sedimentation velocity (AUC-SV), which provides the molecular mass of a protein or protein mixture in solution (Oda et al. Reference Oda, Uchiyama, Noda, Nishi, Koga, Mayanagi, Robinson, Fukui, Kobayashi, Morikawa and Azuma2009; Nishio et al. Reference Nishio, Kamiya, Mizushima, Wakatsuki, Sasakawa, Yamamoto, Uchiyama, Noda, McKay, Fukui, Hauri and Kato2010) in order to exclude the possibility of protein aggregation. C(s) sedimentation coefficient distributions at different concentrations (4·8, 9·6 and 30 μ m) were similar (Fig. 3B), indicating the absence of concentration-dependent interactions. The majority (more than 90%) of TbIMPDH had species at the sedimentation value (s-value) of 14·5 S. The molecular mass of major species at 30 μ m using this s-value and fitted shape factor (f/f 0) is 389 kDa. Although it should be noted that f/f 0 is provided as an averaged value in C(s) analysis and does not correspond to the value for only major species. This molecular mass value of 389 kDa is closer to the molecular mass of 401 kDa calculated from the amino acid sequence, indicating that indeed TbIMPDH is a heptameric protein. We then estimated the molecular mass of species with different s-values by using a Monte Carlo genetic algorithm analysis in which individual s-values, f/f 0 values and populations for different species were obtained by numerical analysis. The estimated molecular mass of major species from Monte Carlo genetic algorithm analyses ranged from 379 to 397 kDa, consistent with a heptameric structure for TbIMPDH (Fig. S1, online version only). The hydrodynamic parameters of the heptamer (s-value of 15·5 S and f/f 0 of 1·2) correspond to an oblate molecule with dimensions of 16·4 nm × 4·9 nm assuming 30% hydration.
Identification of TbIMPDH enzymatic reaction products
In order to confirm that indeed the trypanosomal enzyme is an IMPDH that converts IMP into XMP, we carried out an HPLC analysis of the products resulting from the enzymatic reaction of TbIMPDH. Attempts to identify TbIMPDH-generated XMP revealed that the reaction product resulting from NAD-dependent oxidation reaction of IMP displayed an elution time corresponding to that of the authentic XMP (Fig. 4A). This figure shows also the chromatogram trace of the reaction mixture after incubation at 25 °C for 0 (a), 3 (b) and 25 (c) h. The elution times for NAD and IMP were 2·8 and 7·2 min, respectively, while those of XMP and NADH were 13·6 and 11·8 min, respectively, as compared with the elution times of authentic NAD, IMP, XMP and NADH. Moreover, the oxidation of IMP and biosynthesis of XMP were done in a time-dependent manner (Fig. 4B), confirming an enzyme-catalysed reaction of IMP conversion into XMP. Under our experimental conditions, no other enzymatic reaction products were identified. HPLC analysis results demonstrated that the TbIMPDH-generated product is indeed XMP and thus confirmed the fact that TbIMPDH catalyses specifically the oxidation of IMP into XMP.

Fig. 4. Identification of TbIMPDH products. TbIMPDH (150 nm) was added to a solution containing 250 μ m IMP and 1 mm NAD. (A) After incubation for 0 (a), 3 (b) and 25 (c) h at 25 °C, the reaction mixtures were separated by HPLC with a reversed-phase column at 35 °C. The mobile phase consisted of acetonitrile containing 0·1% trifluoroacetic acid (solvent A) and water containing 5 mm PIC-A reagent (solvent B). The gradient programmer was used according to the following procedure: 0–5 min hold on 5% A, 5–15 min linearly increase A from 5% to 50%, 15–20 min hold on 95% A, 20–30 min hold on 5% A with a flow-rate of 300 μL min−1. The elution was monitored at 254 nm. (B) Consumption of IMP and formation of XMP by TbIMPDH.
Dependence of TbIMPDH activity on pH
As shown in Fig. 5, TbIMPDH exhibited the maximum enzymatic activity at around pH 8·5. However, this activity dropped down rapidly above this pH, suggesting probably that at pH > 8·5, the enzyme may well undergo an alkaline denaturation that would affect its activity. Additionally, to evaluate the alkaline denaturation of TbIMPDH at pH > 8·5, we performed circular dichroism (CD) measurements of TbIMPDH (Fig. S2, online version only). The secondary and ternary structures of protein can be determined by CD spectra in the far- and near-UV region, respectively. The far-UV CD spectra of TbIMPDH were almost identical shapes in the pH range from 7·87 to 9·88, indicating that there was no significant difference in secondary structure. On the other hand, the near-UV CD spectra of TbIMPDH were changed in a pH dependent-manner, suggesting that the ternary structure of aromatic residues are changed in the high pH range and thus TbIMPDH likely undergoes an alkaline denaturation.

Fig. 5. pH dependence of activity for TbIMPDH. Relative activity is plotted vs pH. The plots are normalized by setting the value at pH 8·5 to 100%. Measurements were carried out at 25 °C in the buffers described in the experimental section. The experiment was conducted 3 times at each pH. The values are expressed as the mean±s.e.
Determination of TbIMPDH kinetics parameters
The study of kinetic parameters of TbIMPDH revealed that calculated K m and k cat values for IMP were 30 ± 1·2 μ m and 0·28 ± 0·031 s−1, respectively. Data of the initial velocity versus IMP concentration in the presence of 1·5 mm NAD were collected and fitted to the Michaelis-Menten equation (Fig. 6A). In contrast, the calculated apparent K m and K i values for NAD were 1300 ± 290 μ m and 3·00 ± 0·76 mm, respectively (Table 1) and data of the initial velocity versus concentration of NAD in the presence of 300 μ m IMP were collected and fitted to the uncompetitive substrate inhibition equation (Equation 1, Fig. 6B).

Fig. 6. Steady-state kinetics of TbIMPDH. (A) The plot of initial velocity vs the concentration of IMP at a fixed concentration of NAD (1·5 mm). Data were fitted to a Michaelis–Menten equation. (B) The plot of initial velocity vs the concentration of NAD at a fixed concentration of IMP (300 μ m). Data were fitted to Equation 1, which describes uncompetitive substrate inhibition. The experiment was conducted 3 times at each concentration of substrate. The values are expressed as the mean±s.e.
Table 1. Comparison of the kinetic parameters of IMPDH
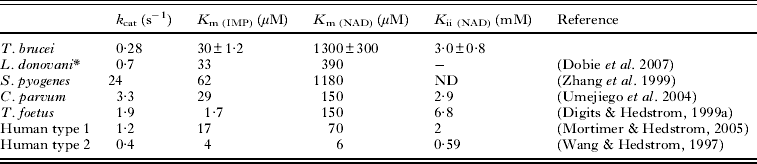
* L. donovani, Leishmania donovani; assays were performed in 100 mm KCl, 50 mm Tris, pH 7·5, 2 mm DTT and 2 mm EDTA at 25 °C.
S. pyogenes, Streptococcus pyogenes; assay condition was not described (Zhang et al. Reference Zhang, Evans, Rotella, Westbrook, Beno, Huberman, Joachimiak and Collart1999).
C. parvum, Cryptosporidium parvum; assays were performed in 100 mm KCl, 50 mm Tris, pH 8·0, 1 mm DTT and 3 mm EDTA at 25 °C.
T. foetus, Tritrichomonas foetus; assays were performed in 100 mm KCl, 50 mm Tris, pH 8·0, 1 mm DTT and 3 mm EDTA at 25 °C.
Human type 1; assays were performed in 100 mm KCl, 50 mm Tris, pH 8·0 and 1 mm DTT at 37 °C.
Human type 2; assays were performed in 100 mm KCl, 50 mm Tris, pH 8·0, 1 mm DTT and 2 mm EDTA at 25 °C.
-, Not detected.
ND, no data.
Kinetics of TbIMPDH inhibitors
We assessed the inhibitory effects of the 3 chemicals on TbIMPDH. Figure 7 shows that RMP, MPA and MZP inhibited TbIMPDH activity in competitive (Fig. 7A), uncompetitive (Fig. 7B) and slow tight competitive inhibition (Fig. 7C) manners, respectively. Calculated K i values for RMP, MPA and MZP were 3·2 ± 0·16 μ m, 21 ± 3·5 nM and 3·3 nM, respectively (Table 2).

Fig. 7. Kinetic analysis of TbIMPDH inhibition. (A) Lineweaver–Burk double-reciprocal plots (1/v vs 1/[IMP]) for TbIMPDH activity are shown in the presence or absence of RMP. A mixture of 200 nm TbIMPDH, 1·5 mm NAD, 10–300 μ m IMP and 0 (open circle), 2 (filled circle), 4·6 (open square), 10 (filled square) or 20 (open triangle) μ m RMP was incubated for 5 min at 25 °C. The data were fitted to the equation describing competitive inhibition. (B) Lineweaver–Burk double-reciprocal plots (1/v vs 1/[NAD]) for TbIMPDH activity are shown in the presence or absence of MPA. A mixture of 200 nM TbIMPDH, 300 μ m IMP, 100–700 μ m NAD and 0 (open circle), 20 (filled circle), 40 (open square) or 60 (filled square) nM MPA was incubated for 5 min at 25 °C. The data were fitted to the equation describing uncompetitive inhibition. (C) Slow tight-binding competitive inhibition by MZP. A mixture of 200 nM TbIMPDH, 160 μ m IMP 1·5 mm NAD and 0, 100, 200, 300, 500 or 750 nM MZP was incubated for 1800 s at 25 °C. The data were fitted to the equation describing slow tight-binding competitive inhibition.
Table 2. Inhibition constants of IMPDH

L. donovani, Leishmania donovani; assays were performed in 100 mm KCl, 50 mm Tris, pH 7·5, 2 mm DTT and 2 mm EDTA at 25 °C.
S. pyogenes, Streptococcus pyogenes; assay conditions were not described in Ref. (Zhang et al. Reference Zhang, Evans, Rotella, Westbrook, Beno, Huberman, Joachimiak and Collart1999).
C. parvum, Cryptosporidium parvum; assays were performed in 100 mm KCl, 50 mm Tris, pH 8·0, 1 mm DTT and 3 mm EDTA at 25 °C.
T. foetus, Tritrichomonas foetus; assays for RMP were performed in 100 mm KCl, 50 mm Tris, pH 8·0 and 1 mm DTT at 25 °C, assays for MPA and MZP were performed in 100 mm KCl, 50 mm Tris, pH 8·0, 1 mm DTT and 3 mm EDTA at 25 °C.
Human type1; assay conditions were not described in Ref. (Hager et al. Reference Hager, Collart, Huberman and Mitchell1995).
Human type2; assay conditions were not described in Ref. (Hager et al. Reference Hager, Collart, Huberman and Mitchell1995).
C, competitive inhibition; UC, uncompetitive inhibition; SC, slow tight competitive inhibition; ND, no data.
DISCUSSION
Human African trypanosomiasis is re-emerging alarmingly in some parts of sub-Saharan Africa. However, the disease has no reliable chemotherapy. Existing drugs are limited in number, present side-effects and drawbacks, and in some cases drug-resistant strains of trypanosomes are emerging. In the past, we have been interested in the identification of new anti-trypanosomal drugs and characterized anti-trypanosomal activities of pro-anthocyanidin from Kola acuminata (Kubata et al. Reference Kubata, Nagamune, Murakami, Merkel, Kabututu, Martin, Kalulu, Huq, Yoshida, Ohnishi-Kameyama, Kinoshita, Duszenko and Urade2005) and artemisinin derivatives from Artemisia annua (Kubata et al. Reference Kubata, Martin and Milhous2008). In Trypanosoma, conversion of IMP to XMP is the major pathway in GMP biosynthesis and thus IMPDH, which catalyses the conversion reaction, is essential for the survival of Trypanosoma. Therefore, we would expect that IMPDH inhibition causes cell death in T. brucei, and a TbIMPDH-specific inhibitor could be an effective anti-trypanosomal drug. So far, Wilson et al. (Reference Wilson, Berens, Sifri and Ullman1994) reported identification of IMPDH mRNA and observation of IMPDH activity in the lysate of T. brucei gambiense. However, there is no other report related to the enzymatic properties of TbIMPDH. In the present study, we cloned and purified the recombinant TbIMPDH by using the E. coli expression system, and carried out the enzymatic characterization of TbIMPDH as a candidate of drug target. Thus, this is a first report for showing the enzymatic characterization of TbIMPDH. Further, we also determined the molecular mass of its denatured (subunit) and native forms. Experimental results showed that TbIMPDH is a very large heptameric protein with almost twice the size of all other tetrameric enzymes described so far. Although it is not yet clear why the trypanosomal enzyme appears as a heptamer, it is tempting to speculate that this structure provides TbIMPDH with additional functional features.
RMP, MPA and MZP are well-known inhibitors of IMPDH from various sources (Patterson and Fernandez-Larsson, Reference Patterson and Fernandez-Larsson1990; Gan et al. Reference Gan, Petsko and Hedstrom2002; Hong and Cameron, Reference Hong and Cameron2002; Gensburger et al.Reference Gensburger, Picard and Marquet2009). Inhibition studies carried out showed that RMP, MPA and MZP were competitive, uncompetitive and slow tight-binding inhibitors, respectively of TbIMPDH as has been described previously. TbIMPDH K i values of 3·2 ± 0·16 μ m, 21 ± 3·5 nM and 3·3 nM, for RMP, MPA and MZP, respectively, are higher than those of mammalian type-enzymes, indicating that these chemicals are stronger inhibitors of mammalian rather than trypanosomal enzymes.
Interestingly, the K m value of 1300 μ m for NAD of TbIMPDH is about 20 to 200-fold higher than that of the mammalian counterpart (K m value of 70 μ m for NAD of human type 1 and K m value of 6 μ m for NAD of human type 2 IMPDHs), showing a difference in the mode and strength of binding of the cofactor NAD by both enzymes. Streptococcus pyogenes IMPDH (SpIMPDH) shows the higher K m value for NAD than human IMPDH and the difference have been explained by the stacking interaction between enzyme and NAD. The K m value of human IMPDH type 2 for NAD (K m = 6 μ m) is lower than that of human IMPDH type 1 for NAD (K m = 70 μ m), and the K m value of human IMPDH type 1 for NAD is lower than that of SpIMPDH (K m = 1180 μ m) (Zhang et al. Reference Zhang, Evans, Rotella, Westbrook, Beno, Huberman, Joachimiak and Collart1999). Colby et al. (Reference Colby, Vanderveen, Strickler, Markham and Goldstein1999) clarified the ternary complex between human IMPDH type 2 and the substrate and cofactor analogues, which are 6-Cl IMP and selenazole-4-carboxamide adenine dinucleotide (SAD). The crystal structure shows that the adenine ring of SAD stacks against the indole ring of His253 and aromatic ring of Phe282 in human IMPDH type 2. In human IMPDH type 1, these residues are replaced by Arg and Tyr, respectively, implying that these substitutions partially disrupt stacking interaction. In SpIMPDH, these residues are replaced by Thr and Gly, respectively, implying that these substitutions completely disrupt the stacking interaction (Fig. 8). The differences between the K m values for NAD correlate with the substitutions of these residues. Thus, we speculated that TbIMPDH has the substitution of these residues such as Thr and Gly of SpIMPDH and the stacking interaction would be disrupted in TbIMPDH. However, these residues were replaced by Arg and Tyr in TbIMPDH, respectively, and similarly in human IMPDH type 1. The K m values of TbIMPDH and human IMPDH type 1 for NAD were 1300 and 70 μ m, respectively. Thus, the stacking interaction is probably unrelated to the high K m value of TbIMPDH for NAD and other factors would influence the NAD binding of TbIMPDH.

Fig. 8. Alignment of the amino acid sequences of the predicted NAD binding site. The primary structure of IMPDHs were aligned by using the CLUSTAL X2 program (Larkin et al. Reference Larkin, Blackshields, Brown, Chenna, McGettigan, McWilliam, Valentin, Wallace, Wilm, Lopez, Thompson, Gibson and Higgins2007). The NCBI ID for sequences used in the sequence alignment analysis were as follows: Leishmania donovani (AAA29253), Streptococcus pyogenes (YP_603464), Cryptosporidium parvum (AAL83208), Tritrichomonas foetus (AAB01581), human Type 1 (NP_000874) and human Type 2 (NP_000875). Identical amino acid residues in at least 4 of 7 sequences are indicated by a grey background. NAD binding residues in human IMPDH type 2 are indicated by ⧫ symbols.
On the other hand, the remarkable difference between the K m values of trypanosomal and mammalian IMPDHs for NAD can most probably be explained by the NAD and NADH regenerating system of glycosome. Vertommen et al. (Reference Vertommen, Van Roy, Szikora, Rider, Michels and Opperdoes2008) reported that TbIMPDH is localized in the glycosome and actually TbIMPDH has a carboxyl-terminal Ser-Lys-Leu tripeptide, which is a glycosomal peroxisomal targeting signal. Glycosome possesses an NAD and NADH regenerating system in trypanosomatids (Michels et al. Reference Michels, Bringaud, Herman and Hannaert2006) and the concentrations of coenzyme in the glycosome would be much higher than in the cytosol. It is considered that TbIMPDH needs a high concentration of NAD to exhibit the enzymatic activity at the high K m value of TbIMPDH for NAD. As described above, TbIMPDH is localized in the glycosome, which possesses a large amount of NAD. Thus, TbIMPDH could exhibit the enzymatic activity in the glycosome, indicating that TbIMPDH adapts to its local environment. A high K m value for NAD has also been reported for the parasitic enzyme Cryptosporidium parvum IMPDH (CpIMPDH) (Umejiego et al. Reference Umejiego, Li, Riera, Hedstrom and Striepen2004), and the NAD binding site of CpIMPDH was also identified as a potential drug target because of the difference between structure and properties of both the parasitic and mammalian enzymes. Actually, benzimidazole derivatives which target the NAD binding site showed a parasite-selective inhibition for CpIMPDH (Kirubakaran et al. Reference Kirubakaran, Gorla, Sharling, Zhang, Liu, Ray, Macpherson, Striepen, Hedstrom and Cuny2012). Therefore, the difference in structure and properties between TbIMPDH and the mammalian enzymes makes TbIMPDH a good candidate for the development of more specific inhibitors targeting its NAD binding site. To achieve this, X-ray crystallographic studies of TbIMPDH and in complex with NAD are indispensable and we are currently working on this.
ACKNOWLEDGEMENTS
We thank Dr K. Fujimori, Ms M. Tabuchi and Ms C. Kimura for technical assistance and helpful discussions. This study was undertaken with the financial support of the Government of Canada provided through the Canadian International Development Agency to NEPAD. This study was also supported in part by the program Grants-in-Aid for Scientific Research of the Ministry of Education, Culture, Sports, Science and Technology of Japan and Osaka Prefecture (grant numbers 17300165, 21500428 and 21200076 (to T.I.)).












