INTRODUCTION
Protozoan parasites of the genus Trypanosoma are widespread and commonly found in all groups of vertebrates. Some are associated with severe clinical disease in humans (T. cruzi, T. brucei gambiense, T. b. rhodesiense) and other animals (T. evansi), and can be particularly detrimental to new or novel host species (Pickering and Norris, Reference Pickering and Norris1996; Wyatt et al. Reference Wyatt, Campos, Gilbert, Kolokotronis, Hynes, DeSalle, Daszak, MacPhee and Greenwood2008). Others are host specific, with little or no apparent negative effects associated with infection, and which appear to rarely become established in cycles involving new hosts (Hoare, Reference Hoare1972; Smith et al. Reference Smith, Telfer, Burthe, Bennett and Begon2005). Except for T. equiperdum, which is a sexually transmitted disease of horses, vertebrate trypanosomes are primarily transmitted by insect vectors, although other blood-sucking invertebrates including ticks and leeches may also play a role in transmission. They are separated into 2 distinct biological groups depending on their developmental pattern in the vector and mode of transmission. The stercorarian trypanosomes (e.g. T. cruzi) develop in the hind gut of an invertebrate vector and are deposited in faeces during feeding, with infection occurring via contamination of the bite wound or mucosal membranes. The second group, the salivarian trypanosomes (e.g. T. brucei), develop in the mid-gut of an insect vector and, following migration to the salivary glands or proboscis, are injected directly into a host during feeding (Hoare, Reference Hoare1972; Brun et al. Reference Brun, Hecker and Lun1998).
While much is known of the trypanosomes that impact on human health or economic development, a great deal less is known of the trypanosomes associated with wildlife. Amongst the unique fauna of Australia, trypanosomes have been reported from numerous vertebrates, including the woylie (Bettongia penicillata), chuditch (Dasyurus geoffroii) (Smith et al. Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008), eastern grey kangaroo (Macropus giganteus), common wombat (Vombatus ursinus), platypus (Ornothorhynchus anatinus) (Noyes et al. Reference Noyes, Stevens, Teixeira, Phelan and Holz1999), long-necked tortoise (Emydura signata) (Jakes et al. Reference Jakes, O'Donoghue and Adlard2001), quokka (Setonix brachyurus) (Clark and Spencer, Reference Clark and Spencer2006), swamp wallaby (Wallabia bicolour) (Hamilton et al. Reference Hamilton, Stevens, Gidley, Holz and Gibson2005 a) and Gilbert's potoroo (Potorous gilbertii) (GenBank Accession no. DQ868978), as well as birds (Mackerras and Mackerras, Reference Mackerras and Mackerras1960) frogs and fish (Mackerras and Mackerras, Reference Mackerras1961) and other mammals (Makerras, Reference Mackerras1959) and reptiles (Mackerras, Reference Mackerras1961). However, those parasites that have been identified from Australian vertebrates may be important for our understanding of both trypanosome evolution and disease processes. For example, Hamilton et al. (Reference Hamilton, Stevens, Gidley, Holz and Gibson2005a) showed that trypanosomes from 3 different marsupial host species in Australia were more closely related to trypanosomes from non-Australian mammals than to each other, suggesting that Australian marsupial trypanosomes have divergent evolutionary origins. Furthermore, it is not yet understood whether Australian trypanosomes have a negative impact on the host, and therefore whether they should be a concern for conservation initiatives (Smith et al. Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008; Department of Environment and Conservation, 2008).
Here we report an investigation into the diversity of trypanosomes associated with Australian wildlife based on divergence of the 18S rRNA gene. We also report on the diversity of trypanosomes within selected host species sampled across large geographical areas and varied environments, including mainland and offshore island populations, in an attempt to determine whether Australian trypanosomes are host-specific, or if there is regional variation in host-parasite associations. The 18S rRNA gene was targeted as it is generally conserved throughout the eukaryotes, but due to different rates of evolution among different regions within the gene, it can be used to infer relationships across a wide phylogenetic range (Sogin et al. 1986).
Materials and Methods
Collection and preparation of samples
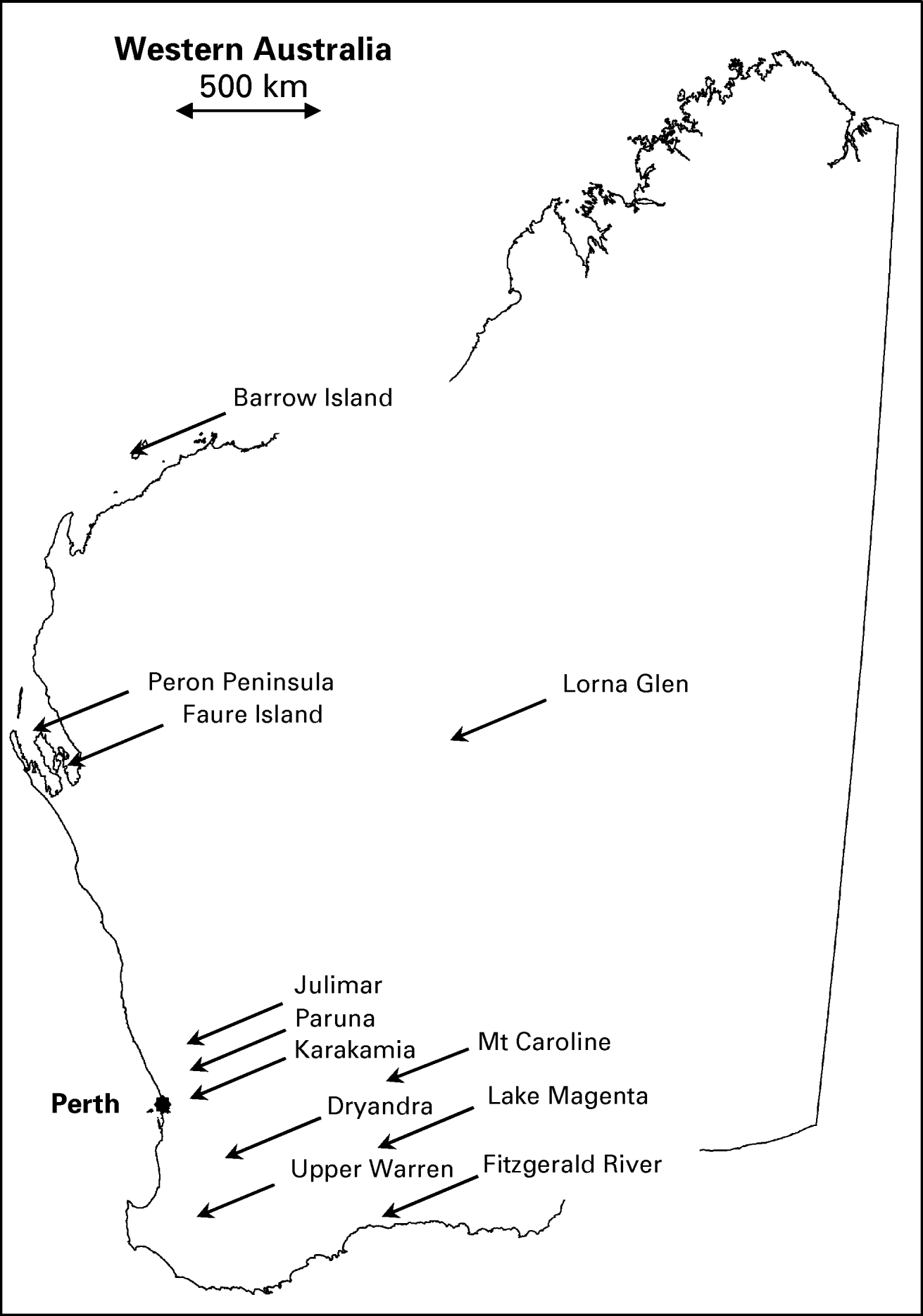
Blood samples were obtained from 371 native mammals of 19 different species, trapped from 14 different locations throughout Australia (12 within Western Australia) as part of the Western Australian Department of Environment and Conservation (DEC) fauna research and monitoring programs between March 2007–August 2008 (Table 1, Fig. 1). All animals were live-trapped using either Sheffield wire-cages or Elliot traps baited with a mixture of peanut butter, rolled oats and sardines. Blood samples ranged in size from 50 to 500 μl depending on the species and were obtained from either the lateral caudal vein (boodie Bettongia lesueur; woylie; brush-tailed possum Trichosurus vulpecula), marginal ear vein (chuditch; bandicoot Isoodon auratus), or dorsal pedal vein (Planigale sp.; Shark Bay mouse Pseudomys fieldi; bush rat Rattus fuscipes; ash grey mouse Pseudomys albocinereus; dibbler Parantechinus apicalis). Whole blood was collected in 1·5 ml Eppendorf tubes and centrifuged within 6 h at 10 600 g for 10 min to separate and remove sera. The resulting blood pellets were stored at −20°C. Genomic DNA was extracted from blood using the QIAamp Blood DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Wildlife sampling was carried out under Murdoch University Animal Ethics Approval Permit Numbers NS1182-06 and W2172-08, and DEC Animal Ethics Approval Permit Number DEC AEC/21/2008.
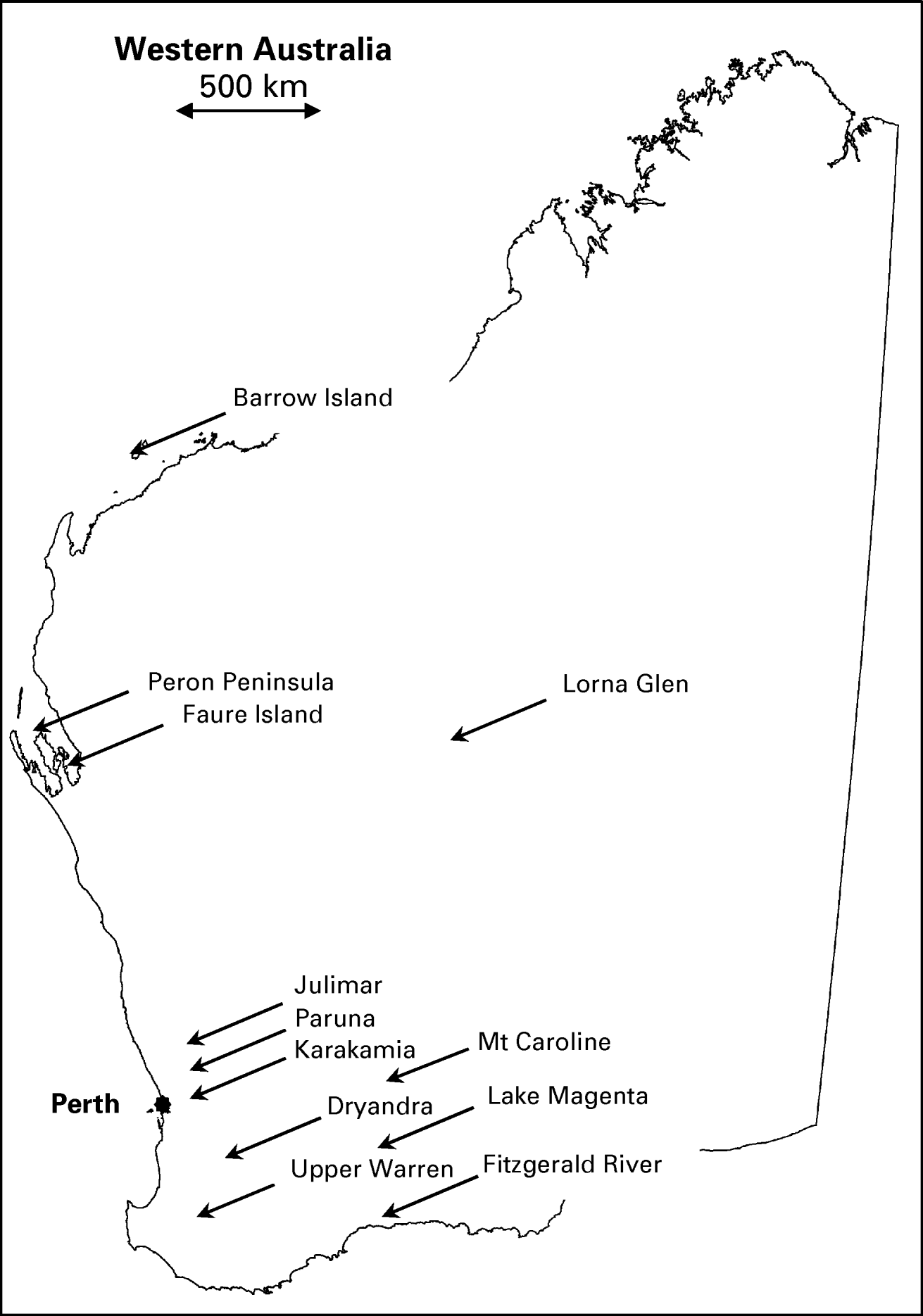
Fig. 1. Map of Western Australia showing approximate locations of the 12 sampling sites in relation to Perth city (filled circle). Sampling sites in New South Wales (Scotia) and South Australia (Thistle Island) are not shown.
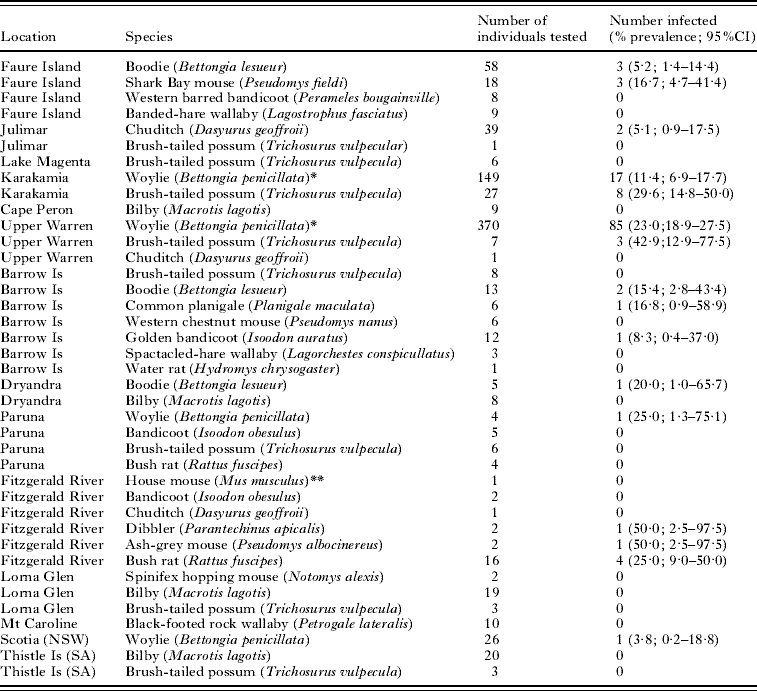
Table 1. Geographical location within Western Australia, host species, sample size and trypanosome infection prevalence (%) of samples collected during the present investigation
(* Indicates data published previously by Smith et al. (Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008). ** Indicates non-native species. NSW, New South Wales; SA, South Australia.)

Detection and amplification of the 18S rRNA gene
A sample of 10 μl of DNA extract was used as a template in a nested PCR using trypanosome-specific oligonucleotides as described by Noyes et al. (Reference Noyes, Stevens, Teixeira, Phelan and Holz1999). In the first round, each 50 μl reaction also contained 5 μl of 10× buffer (20 mm Tris-HCl at pH 8·8; 10 mm KCl; 10 mm (NH4)2SO4; 2 mm MgSO4; 0·1% Triton® X-100 and 0·1 mg/ml nuclease-free BSA); 200 μm dNTPs (Promega, Madison, USA); 1 U Taq DNA polymerase (Promega, Madison, USA); 0·5 U of the proof-reading enzyme Pfu (Promega, Madison, USA) and 8 pmol of the following external primers: TRY927F 5′ GAAACAAGAAACACGGGAG 3′, TRY927R 5′ CTACTGGGCAGCTTGGA 3′, for 30 cycles of 94°C 30 s; 55°C 60 s; 72°C 90 s. The products from the first amplification were diluted 1:10 in sterile deionized water. Then 2 μl of the product from each reaction was used as a template for the second-round PCR using the following internal primers: SSU561F 5′ TGGGATAACAAAGGAGCA 3′, SSU561R 5′ CTGAGACTGTAACCTCAAAGC 3′ and the same cycling conditions described above. Amplified products were separated on a 1·5% agarose gel and run at 100 V for 1 h in Tris-acetate-EDTA buffer and visualized under UV light. Samples producing a band of approximately 600 bp were considered positive. Prevalence of parasitic infection was expressed as the percentage of hosts with a positive DNA sample and 95% confidence intervals were calculated assuming a binomial distribution, using the software Quantitative Parasitology 3.0 (Rózsa et al. Reference Rózsa, Reiczigel and Majoros2000).
Sequencing and phylogenetic analysis of Australian trypanosomes
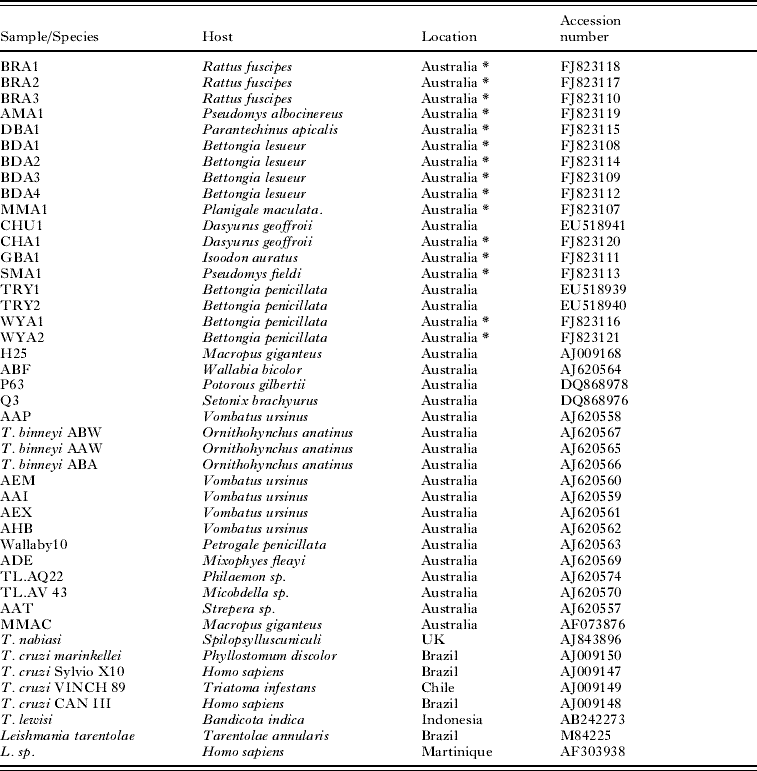
PCR products were purified using Wizard SV PCR and gel purification kit (Promega, Madison, USA) according to the manufacturer's instructions. Sequencing reactions were initiated using the purified PCR products as templates with the following cycling conditions: initial cycle at 96°C for 120 s followed by 25 cycles of 96°C for 10 s, 54°C for 5 s and 60°C for 240 s. Sequences were analysed with FinchTV™ (Geospiza Inc, USA). Construction of the phylogenetic tree was carried out by comparing the resulting 18S rRNA sequences with known sequences in the GenBank database using ClustalW (Higgins et al. Reference Higgins, Thompson, Gibson, Thompson, Higgins and Gibson1994). The host species, geographical origin and Accession number of all sequences used in the phylogenetic analyses are shown in Table 2. In order to acquire a better understanding of the phylogeny of Australian trypanosomes, the sequence alignment included representatives of a major disease-causing species of trypanosomes, T. cruzi. Phylogeny was determined by neighbour-joining, minimum evolution and maximum parsimony methods, implemented with MEGA Version 4.0 (Tamura et al. Reference Tamura, Dudley, Nei and Kumar2007). The robustness of branches within the resulting trees was tested by 100 cycles of bootstrap resampling. For distance estimation methods, evolutionary distances were computed using the maximum composite likelihood method. All positions containing gaps and missing data were eliminated from the dataset. There was a total of 77 positions in the final dataset. The trypanosomatid genera Leishmania was used as an outgroup to root the tree.
Table 2. Trypanosoma species or isolate code, host species, geographical location and GenBank Accession number of the 44 genotypes included in the current analysis
(Novel isolates detected during the present investigation are indicated by *.)

Phylogenetic analysis of T. lewisi-like trypanosomes
For phylogenetic analysis of the closely related T. lewisi-like trypanosomes obtained in this study, BRA1, BRA3, AMA1 and DBA1 sequences were aligned with other trypanosome 18S rRNA sequences in the GenBank database. The alignment was carried out with ClustalW followed by manual alignment. Phylogeny was determined by neighbour-joining, minimum evolution and maximum parsimony methods, implemented with MEGA Version 4.0. The robustness of branches within the resulting trees was tested by 100 cycles of bootstrap resampling. For distance estimation methods, evolutionary distances were computed using the maximum composite likelihood method. All positions containing gaps and missing data were eliminated from the dataset. There was a total of 444 positions in the final dataset. A consensus was made of the shortest trees.
RESULTS
Trypanosomes were found in 9 of the 19 native mammal species that were trapped in this study. Trypanosomes have been recorded previously in the woylie and chuditch (Smith et al. Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008), but this is the first record for the boodie, brush-tailed possum, golden bandicoot, Shark Bay mouse, bush rat, ash-grey mouse, dibbler and common planigale (Planigale maculata).
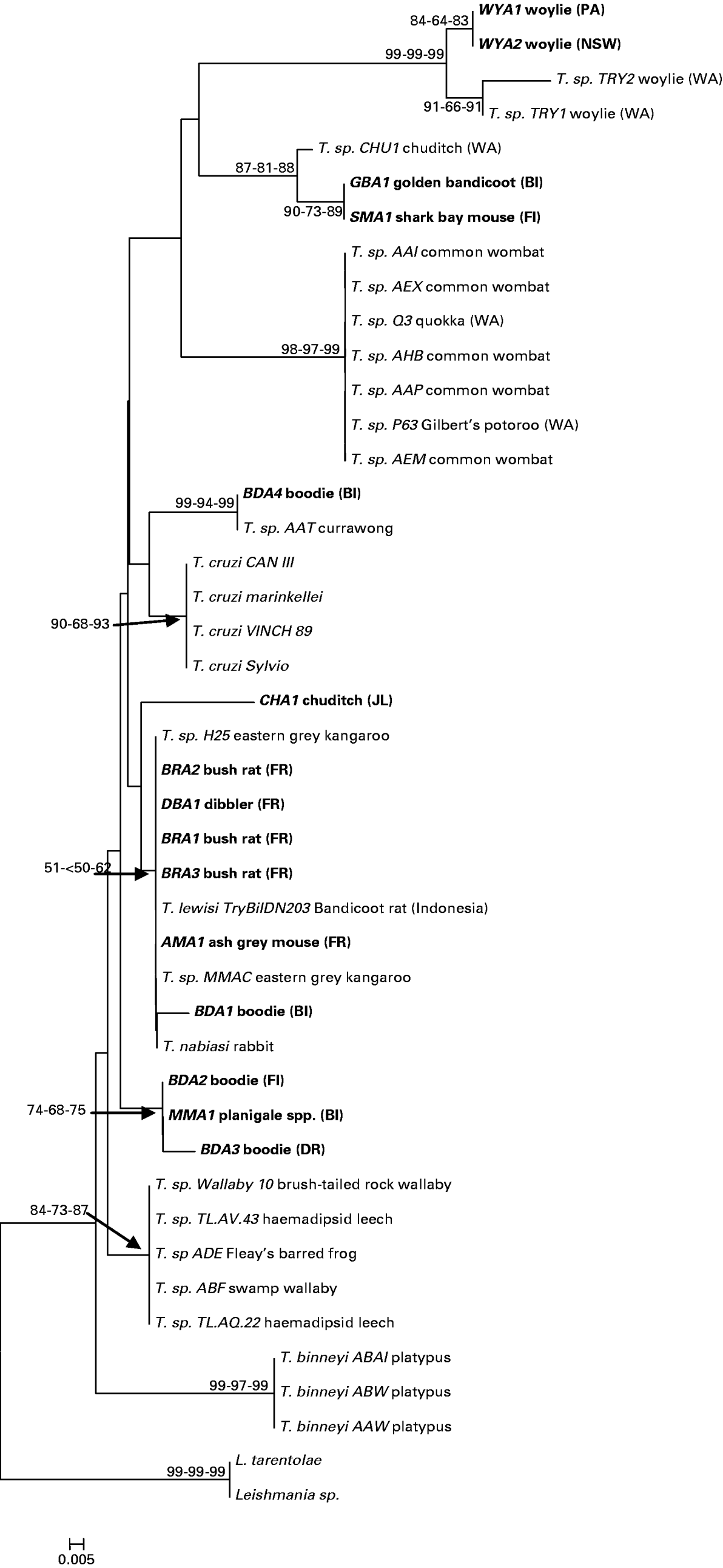
The partial 18S rRNA genes from Australian trypanosomes sequenced in this study were compared with data available in GenBank and showed high homology with 18S rRNA of other Trypanosoma spp. (Table 3). The neighbour-joining tree of evolutionary relationships among the 18S rRNA sequences produced a number of strongly supported clades, with similar support inferred using both maximum parsimony and minimum evolution (Fig. 2). A total of 13 novel genotypes were detected; 4 in the boodie, 2 in the woylie, 2 in the bush rat, 1 in the chuditch, common planigale, ash-grey mouse and dibbler, and a single genotype that was detected in both the Shark Bay mouse and golden bandicoot.
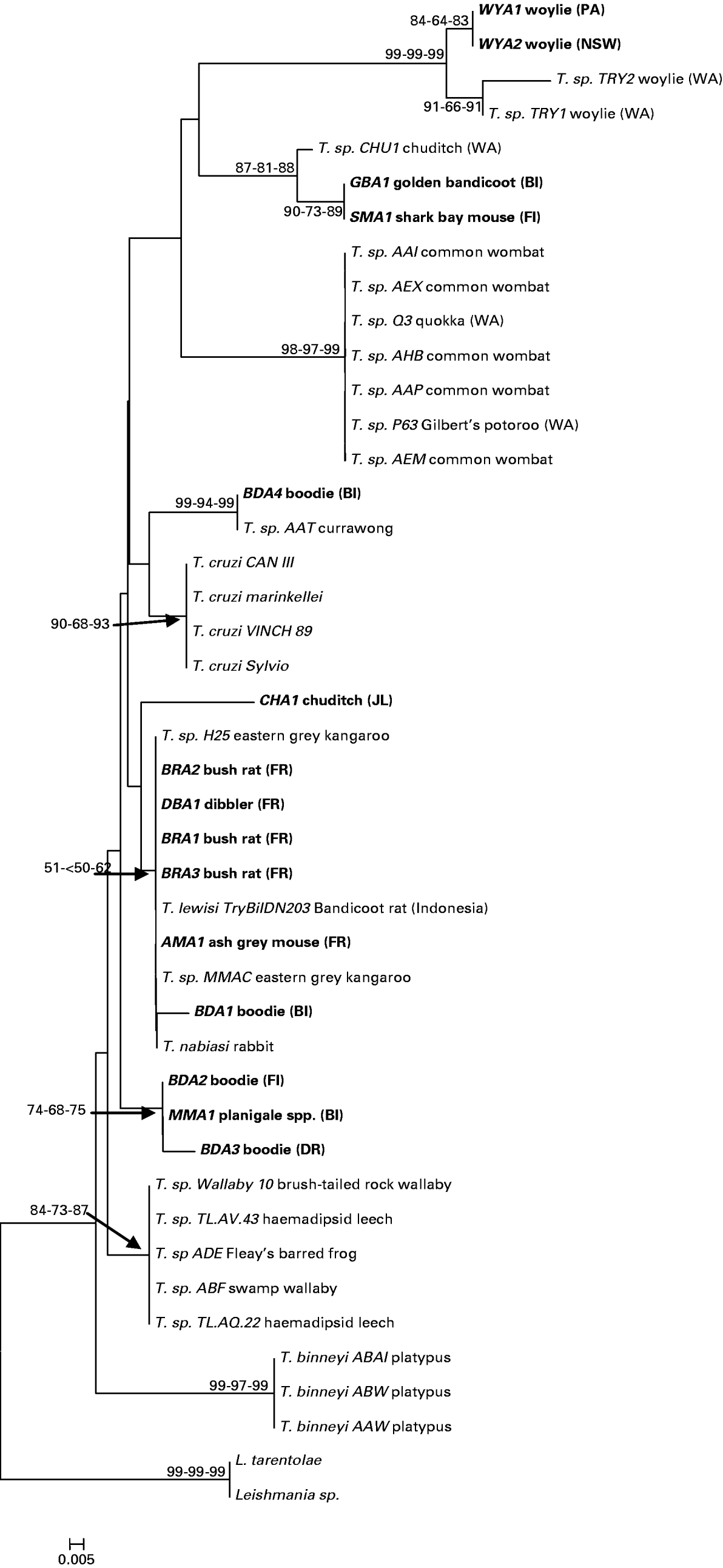
Fig. 2. Phylogram showing the relationship between 44 taxa at the 18S rRNA gene. The relationship was inferred using the neighbour-joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (100 replicates) is shown next to the branches or indicated by arrows. The distances for neighbour-joining analysis were computed using the maximum composite likelihood method. Multiple bootstrap values at nodes are in order: NJ, MP, ME. All positions containing gaps and missing data were eliminated from the dataset. There was a total of 77 positions in the final dataset. Phylogenetic analyses were conducted in MEGA4. Clades without bootstrap values have bootstrap values of <50%. Host species are indicated after Trypanosoma species or isolate code. Genotypes sequenced during this study are shown in bold. The letters in parentheses refer to sampling locality: BI Barrow Island; FR Fitzgerald River National Park;.JI Julimar Conservation Park; DR Dryandra: FI Faure Island; PA Paruna; NSW New South Wales (see Fig. 1). Other Western Australian genotypes reported previously are indicated as WA.
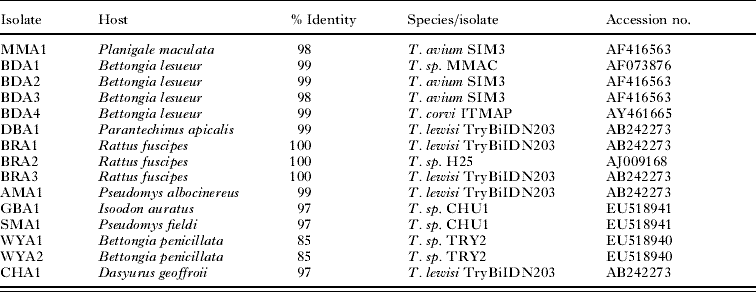
Table 3. Isolate code and host as determined during the current investigation, with the % identity and accession details of the most similar Trypanosoma genotype registered on GenBank as determined by BLAST analysis

Trypanosome sequences from 3 woylie populations in the south west of Western Australia, and 1 population in Scotia Reserve in New South Wales, form a distinct subclade (WYA1, WYA2, TRY1 and TRY2). Two genotypes obtained from bush rats within the Fitzgerald River National Park (BRA1 and BRA3) were identical to each other and showed 100% identity to T. lewisi, a widely distributed trypanosome usually associated with the black rat (R. rattus). Two additional genotypes, dibbler (DBA1) and ash-grey mouse (AMA1) showed 99% identity with T. lewisi and also grouped with other T. lewisi-like trypanosomes (Table 3, Fig. 2). The similarity of these 4 genotypes across the variable V7–V8 region is given in Table 4, and their relationship to other T. lewisi-like trypanosomes from outside Australia (Hamilton et al. Reference Hamilton, Stevens, Holz, Boag, Cooke and Gibson2005b) in Fig. 3.
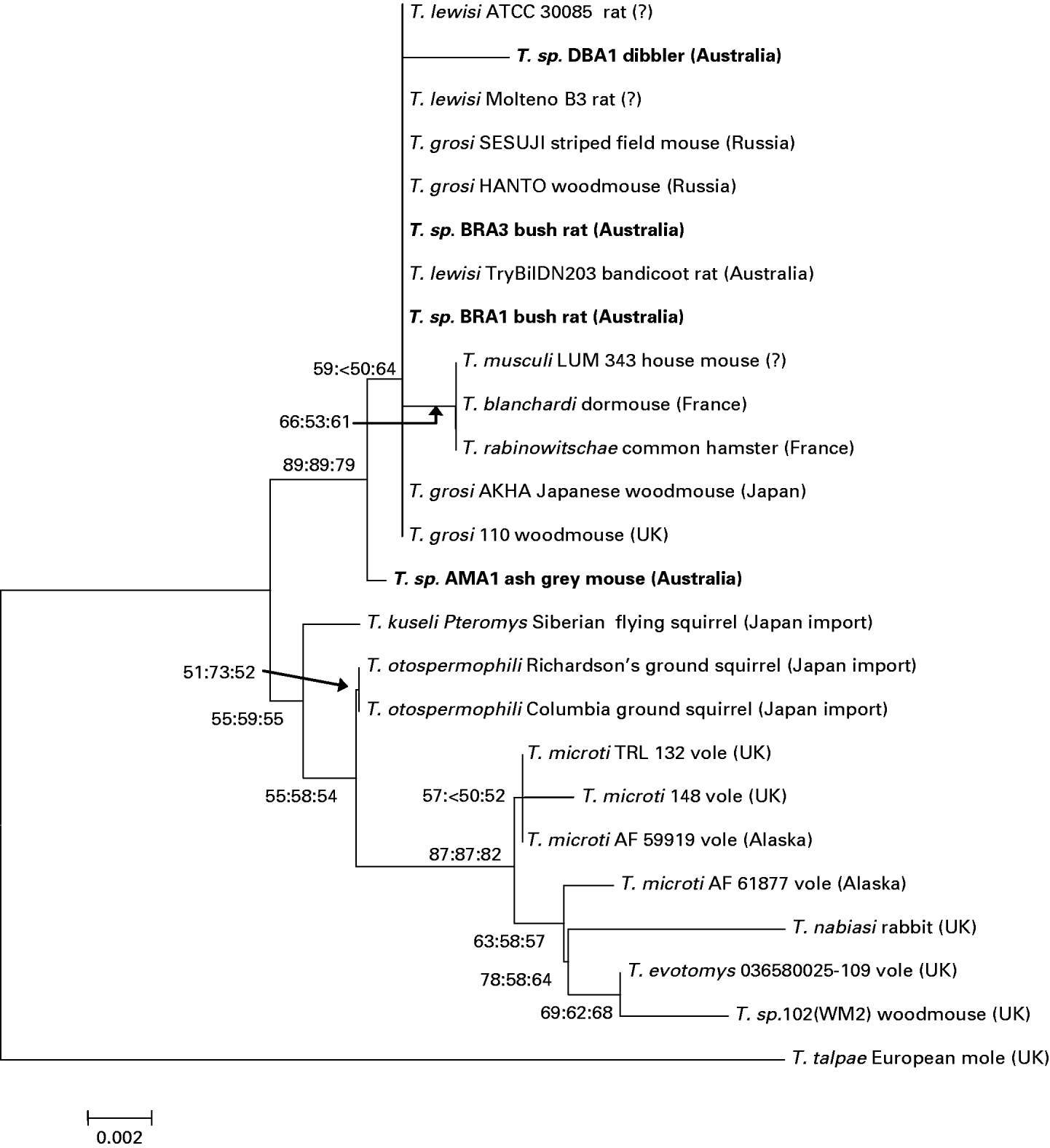
Fig. 3. Phylogram showing the relationship between Trypanosoma lewisi-like genotypes (DBA1, AMA1, BRA1 and BRA3) isolated during the current investigation and those from regions outside Australia after Hamilton et al. (Reference Hamilton, Stevens, Holz, Boag, Cooke and Gibson2005a) inferred using the Neighbour-Joining method. The bootstrap consensus tree inferred from 100 replicates is taken to represent evolutionary history of the taxa. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. Multiple bootstrap values at nodes are in order: NJ, MP, ME. Codon positions included were 1st+2nd+3rd+Noncoding. All positions containing gaps and missing data were eliminated from the dataset (Complete Deletion option). There was a total of 444 positions in the final dataset. Phylogenetic analyses were conducted in MEGA4. Host species, isolate code and country of origin are indicated for each sequence. Genotypes sequenced during this study are shown in bold.
Table 4. Comparison of Trypanosoma lewisi-like nucleotide sequences obtained during this study with T. lewisi from an Indonesian bandicoot rat
(Gaps and regions of alignment between each sequence and T. lewisi are shown.)

Other 18S rRNA sequences showed little congruence with either host or geographical distribution (Fig. 2). Trypanosoma genotypes from a common planigale (MMA1), and 2 boodies (BDA2 and BDA3) grouped together on 1 dichotomous branch with the other branch occupied by all other Australian genotypes isolated in this study. The boodie and common planigale genotypes, although closely related, originate in 3 different geographical locations, Faure Island, Barrow Island and Dryandra on the mainland. In contrast, genotype BDA4, also from a Barrow Island boodie, appears distantly related and forms a cluster with an isolate from an Australian currawong (Strepera spp.), whilst the other isolate from a Barrow Island boodie (BDA1) falls within the T. lewisi group. The chuditch genotype CHA1 showed 97% homology to T. lewisi, whereas an isolate reported previously by Smith et al. (Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008) from the same population (CHU1) appears distinct and more similar to T. benetti isolated from an American falcon.
DISCUSSION
The main purpose of our study was to assess the diversity of trypanosomes associated with a range of Australian (particularly Western Australian) wildlife, and to search for patterns of host and geographical specificity. The overall pattern appears to be one of widespread distribution, with trypanosome genotypes occurring in many different host species, often at high prevalences, across the state as well as on offshore islands.
Although we have not found multiple infections within any single individual host, there is evidence to suggest that some species can be infected by multiple genotypes within the same location, for example 2 distinct genotypes occur in chuditch within Julimar Conservation Park. There is also evidence to suggest that some genotypes are widespread and occur in multiple host species, while others are more likely to be associated with certain hosts or within certain geographical areas. For example, isolates detected in woylies from geographically different populations, including a population in New South Wales, consistently group together within a single clade. Those from the closely related boodie appear not only to be genetically different from genotypes from woylies, but to vary considerably both within and between geographically distinct populations, suggesting that some host species may be susceptible to infection from multiple Trypanosoma spp. and therefore possibly susceptible to exotic species should they be introduced into Australia. Furthermore, based on the section of 18S rRNA gene sequenced, it also appears that several host species within the Fitzgerald River area, including the bush rat, dibbler and ash-grey mouse, were all infected with a single or closely related T. lewisi-like group of parasites, suggesting there may be areas where specific trypanosomes, or their vectors, are more prevalent. Our results repeatedly position the kangaroo isolate H25 within the T. lewisi-like clade, albeit with relatively low bootstrap support, whereas it has previously been positioned within the T. cruzi group (Smith et al. Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008; Stevens et al. Reference Stevens, Noyes, Dover and Gibson1999). This may be an artefact of the single gene region that we were able to sequence or it may be due to the increased number of Australian taxa in the T. lewisi clade, which was the closest group to the T. cruzi clade in previous analyses (Hamilton et al. Reference Hamilton, Stevens, Gidley, Holz and Gibson2005a).
We recognize the limitations of using only the 18S rRNA locus and the need to target additional genes to specifically address the phylogenetic re-positioning of the H25 isolate and the relationships between isolates from native wildlife and T. lewisi. Considerable effort was put into the amplification of both the internal transcribed spacer (ITS) and glyceraldehyde phosphate dehydrogenase (GAPDH) genes without success. This was principally due to logistic constraints of sampling in remote areas over long periods of time and often in extremely high temperatures.
The wide geographical distribution of several genotypes seen in our study may have been influenced by the relatively recent translocation of endangered species within Western Australia. For example, boodie populations on Faure Island and Dryandra were both originally sourced from Dore Island, 1 of only 3 extant wild populations, suggesting a potentially similar geographical origin for their associated parasites. The wild population of boodies on Barrow Island, however, have been isolated from the mainland for approximately 8000 years (Dortch and Morse, Reference Dortch and Morse1984), so it is surprising to note the similarity between genotypes originating there with genotypes obtained from bush rats in Fitzgerald River on the south coast of Western Australia and a kangaroo from Victoria.
Recent phylogenetic studies based on the 18S rRNA gene have identified extensive genetic variation within in the genus Trypanosoma (Stevens et al. Reference Stevens, Noyes, Schofield and Gibson2001). The diversity of genotypes presented here suggests that this is also true of trypanosomes associated with Australian wildlife. The general lack of host specificity also supports the conclusions of Hamilton et al. (Reference Hamilton, Gibson and Stevens2007) who suggested that co-speciation in association with vertebrate hosts appears to have played little or no role in the evolution of trypanosome diversity. However, the fact that we do not find a single genotype infecting all cohabiting hosts in any one area indicates that there are factors maintaining a degree of host specificity. This may well be due to the association of different vectors with different host species.
The vectors of Australian trypanosomes are not known, although Noyes et al. (Reference Noyes, Stevens, Teixeira, Phelan and Holz1999) and Hamilton et al. (Reference Hamilton, Stevens, Gidley, Holz and Gibson2005a) have proposed that haemadipsid (jawed) leeches could act as vectors of some mammalian Trypanosoma spp. in Australasia, as they do elsewhere. Jakes et al. (Reference Jakes, O'Donoghue and Adlard2001) have proposed tabanid flies as possible vectors for kangaroo trypanosomes, although ixodid ticks are also commonly found on kangaroos and are thought to be involved in the transmission of trypanosomes in Japan (Thekisoe et al. Reference Thekisoe, Honda, Fujita, Battsetseg, Hatta, Fujisaki, Sugimoto and Inoue2007). Phylogenetic grouping of genotypes from several ecologically similar wildlife hosts including bush rat, dibbler, and ash-grey mouse, together with the flea-transmitted T. lewisi, a known stercorarian Trypanosoma sp., suggests a potentially similar mode of transmission (Hamilton et al. Reference Hamilton, Gibson and Stevens2007). However, ecological similarity of hosts does not explain the positioning of the genotype isolated from a Barrow Island boodie within the T. lewisi-like group, as they are both geographically isolated and live in large communal burrow systems similar to rabbit warrens.
It is now well accepted that parasites and pathogens can have a significant impact on wildlife population dynamics (Hudson et al. Reference Hudson, Dobson and Newborn1992, 1998; Tompkins and Begon, Reference Tompkins and Begon1999; Albon et al. Reference Albon, Stien, Irvine, Langvatn, Ropstad and Halvorsen2002; Newey and Thirgood, Reference Newey and Thirgood2004; Hawlena et al. Reference Hawlena, Bashary, Abramsky and Krasnov2007; Møller and Nielsen, Reference Møller and Nielsen2007; Pedersen et al. Reference Pedersen, Jones, Nunn and Altizer2007; Burthe et al. Reference Burthe, Telfer, Begon, Bennett, Smith and Lambin2008). Parasites that infect a new or naïve host can be particularly pathogenic due to the absence of an appropriate immune response. T. lewisi, usually considered to be a non-pathogenic trypanosome, has been shown to vary significantly and often unpredictably in virulence and resultant clinical impact in rats, with higher incidences of anaemia and weight loss, and significantly greater mortality rates in juveniles than adults (Brown, Reference Brown1914, Reference Brown1915). In addition, the virulence of non-pathogenic strains has been shown experimentally to increase following passage through cold-blooded animals (Wendelstadt and Fellmer, 1909, cited by Brown, Reference Brown1914). Wyatt et al. (Reference Wyatt, Campos, Gilbert, Kolokotronis, Hynes, DeSalle, Daszak, MacPhee and Greenwood2008) also presented evidence that the extinction of the Christmas Island rat (Rattus macleari) was caused by T. lewisi, which was inadvertently introduced onto the island by ship rats (R. rattus) c1900.
T. lewisi was first reported in Australia by Bancroft (Reference Bancroft1888) and was subsequently reported in bush rats and water rats (Hydromys chrysogaster) (Mackerras, Reference Mackerras1959). Its presence within native wildlife raises questions regarding its origin, specifically whether it was introduced along with black and brown rats on ships arriving from Europe or was present before their arrival. It also raises questions regarding the impact of infection on other native wildlife species in terms of apparent plasticity in virulence. For instance, the variants reported here may reflect a recent evolutionary adaptation by T. lewisi to new hosts, a process that could result in the development of increased virrulence on contact with either the original or additional new hosts (Brown, Reference Brown1914). The sudden emergence of a virulent form of T. lewisi could conceivably result in localized epidemics amongst naïve host species with potentially detrimental effects, although there is no direct evidence to support this at present. Furthermore, virulence of trypanosome infection is known to vary depending on the presence of concomitant infections and the general condition of the host (Cox, Reference Cox2001; Brown et al. Reference Brown, Loosli and Schmid-Hempel2000). For example, Smith et al. (Reference Smith, Clark, Averis, Lymbery, Wayne, Morris and Thompson2008) reported a higher prevalence and intensity of trypanosome infection in a declining population of woylies compared to that observed in a stable non-declining population in the southwest of Western Australia. They suggested that the impact of infection was only of significance for animals in the declining population because they were also exposed to high levels of the parasite Toxoplasma gondii (Arrea et al. Reference Arrea, Carmona, Bermudez and Abrahams1998). The physiological stress caused by the presence of introduced predators such as cats and foxes, as well as stress resulting from poor nutrition, competition for food and resources, and the effects of habitat degradation, are all thought to contribute to both host susceptibility and severity of infection (McCallum and Dobson, Reference McCallum and Dobson2002; Davey et al. Reference Davey, Sinclair, Pech, Arthur, Krebs, Newsome, Hik, Molsher and Allcock2006; Pedersen and Grieves, Reference Pedersen and Grieves2008).
A more comprehensive understanding of the diversity of Trypanosoma spp. associated with native wildlife will contribute to the conservation efforts and translocation programmes of endangered species in Western Australia. Assessment of the current range of trypanosome infections will also allow inferences to be made regarding the susceptibility of host species to the introduction of non-native pathogenic trypanosomes, such as T. evansi, thus helping to guide biosecurity initiatives (Thompson et al. Reference Thompson, Owen, Puana, Banks, Davis and Reid2003). Furthermore, it is important to understand the current range of infections that exist within wildlife populations to be able to predict their suitability to act as competent reservoirs for introduced zoonoses in the event that climate change (Polley and Thompson, Reference Polley and Thompson2009) should result in Australia becoming a suitable environment for establishment (Thompson et al. Reference Thompson, Kutz and Smith2009). As in other parts of the world, increasing encroachment of humans on wildlife habitats enhances the opportunity for novel pathogens to spread to humans, their livestock and pets.
We would like to thank staff at the Australian Wildlife Conservancy and the Department of Environment and Conservation, and Halina Burmej from Murdoch University, for assistance in collecting and processing field samples and for providing access to field sites, and to Nicole Willers for supplying P. lateralis samples. This work was funded by the Australian Research Council and the Department of Environment and Conservation under ARC-Linkage Project ID: LP0775356.