Introduction
Nymphaea tetragona represents one of the naturally occurring species found in India (Mitra, Reference Mitra, Nayar, Thothathri and Sanjappa1990). Its distribution is restricted to one particular location found at Nongkrem, Meghalaya, India (25°28′N–91°52′E). It is considered to be rare and endangered and has been included for recovery programmes (Ganeshaiah, Reference Ganeshaiah2005). Besides N. tetragona, there are an additional nine species (both wild and cultivated) of the genus Nymphaea occurring in India (Mitra, Reference Mitra, Nayar, Thothathri and Sanjappa1990). However, traditional classification of the genus in India has received unconvincing treatments with some names inaccurately used (Cook, Reference Cook1996). To vindicate Cook's remarks, molecular taxonomic revision of four Indian representatives of the genus Nymphaea viz. N. nouchali, N. pubescens, N. rubra and N. tetragona based on internal transcribed spacers (ITS), trnK intron and matK gene was reported (Dkhar et al., Reference Dkhar, Kumaria, Rama Rao and Tandon2010). Molecular evidence suggested probable misidentification of one specimen of N. nouchali and N. tetragona. Furthermore, a comparison of the genetic closeness among the Indian, Chinese and Russian material of N. tetragona based on the ITS region was also evaluated. The results indicated a close relationship between the Indian and the Chinese representatives, which is further supported by the morphological similarities shared between them (eFloras, Reference eFloras2008).
An interesting observation made from the previous study was the relatively higher number of nonsynonymous substitutions as compared to synonymous substitutions detected in the matK gene of N. tetragona. The higher rate of nonsynonymous substitutions is usually inferred as an indication of selective pressures acting on protein-coding sequences. The chloroplast matK gene, coding for the maturase enzyme, has been reported to evolve under positive selection in some lineages of land plants (Hao et al., Reference Hao, Chen and Xiao2010). Hao et al. (Reference Hao, Chen and Xiao2010) showed that several regions (N-terminal region, RT domain and domain X) of matK experience molecular adaptation, which fine-tunes maturase performance.
In the present study, to gain insight into whether an adaptive evolutionary process is linked to the colonization of new habitats by N. tetragona, we tested whether there was selective pressure in matK using phylogenetic and maximum likelihood analyses of codon and branch site substitution models. This estimation was extended to members of the order Nymphaeales, not included in the study of Hao et al. (Reference Hao, Chen and Xiao2010), representing a lineage at the base of the angiospermic tree.
Materials and methods
Taxon sampling and nucleotide sequence data
Sequence data of matK gene and trnK intron of four wild Indian representatives of the genus Nymphaea, viz. N. nouchali, N. pubescens, N. rubra and N. tetragona was taken from our own previous study (Dkhar et al., Reference Dkhar, Kumaria, Rama Rao and Tandon2010). The rbcL gene was amplified and sequenced following the method adopted in another study (Dkhar et al., in press). In addition, barring Nymphaea, nucleotide sequence data of members of the order Nymphaeales (Barclaya longifolia, Euryale ferox, Nuphar advena, Ondinea purpurea and Victoria cruziana), Amborella trichopoda and Austrobaileya scandens, identified as the most basal angiosperms, were retrieved from GenBank.
Molecular evolutionary analysis
In order to conduct phylogenetic maximum likelihood analysis of selection in chloroplast genes, a phylogenetic tree was reconstructed based on the chloroplast trnK intron, matK and rbcL genes using maximum likelihood method implemented in Phylip (Felsentein, Reference Felsentein1989).
Molecular adaptation tests on the chloroplast matK codon sites were performed using maximum likelihood models and programs included in PAML ver. 4.3 (Yang, Reference Yang2007). We performed five site-specific codon substitution models: null models for testing positive selection (M1A, M7 and M8A) and models allowing for positive selection (M2A and M8). Furthermore, to provide evidence whether positive selection is linked to the colonization of N. tetragona, the branch site models (model A with ω fixed or estimated) implemented in PAML ver 4.3 were used. The likelihood ratio test (LRT) was used to compare these alternative models.
Results and discussion
Positive selection in matK gene
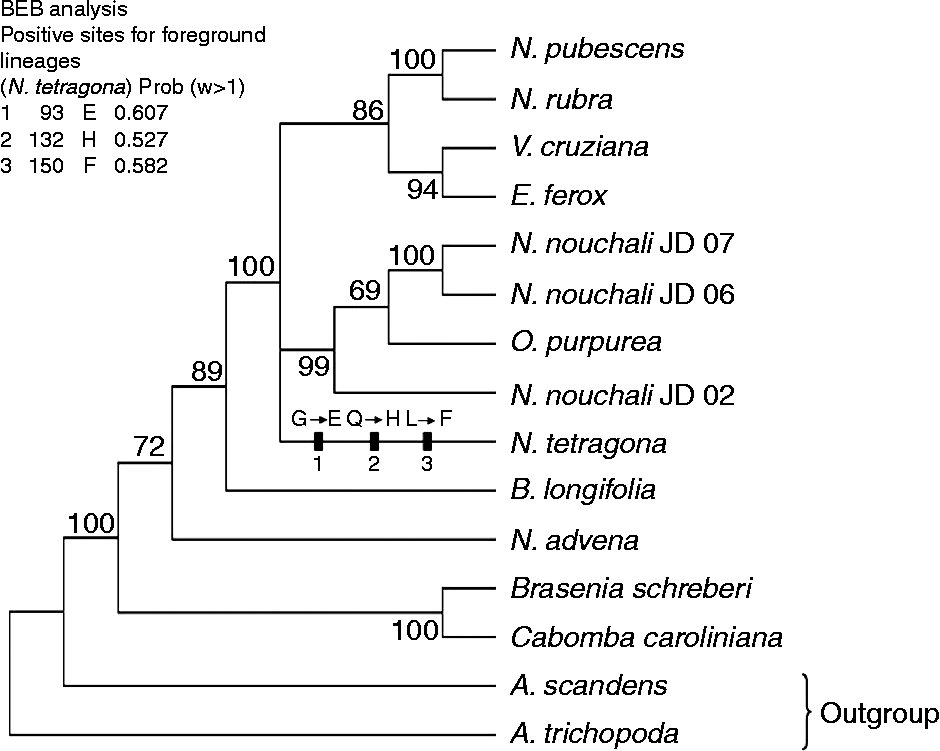
Maximum likelihood analysis of the combined datasets yielded a phylogenetic tree with log likelihood value of − 10 921.95 (Fig. 1). The reconstructed phylogenetic tree was identical in topology to those obtained in a study by Löhne et al. (Reference Löhne, Borsch and Wiersema2007).

Fig. 1 Maximum likelihood tree of combined chloroplast DNA dataset (3859 bp) using Phylip. Three positive sites with probability for foreground (N. tetragona) estimated using branch-site models of PAML are indicated. Amino-acid replacements at these sites are also indicated at the branch (G: Glycine, E: Glutamic acid, Q: Glutamine, H: Histidine, L: Leucine, F: Phenylalanine). Numbers at nodes indicate bootstrap values. Prob, probability.
For detecting positive selection using codon substitution models, three LRTs were performed comparing M1A (nearly neutral) with M2A (positive selection), M7 (β) with M8 (β and ωs ≥ 1), and M8A (β and ωs = 1) with M8 (β and ωs ≥ 1). Because positive selection operates more strongly at some sites (Aguileta et al., Reference Aguileta, Refrégier, Yockteng, Fournier and Giraud2009), the distribution of sites influenced by selection for all three regions (N-terminal region, RT domain and domain X) of the matK gene was evaluated. For N-terminal region, model M2A indicated that 13.2% of the sites are under positive selection (ω = 1.778). In model M8, 13.0% of the sites are under selective pressure (ω = 1.791). For RT domain, model M8 estimated an ω value of 46.57 for 0.01% of codon sites. No positive sites were detected for domain X, a relatively more conserved and important functionally, as it determines the maturase activity of matK proteins (Hausner et al., Reference Hausner, Olson, Simon, Johnson, Sanders, Karol, McCourt and Zimmerly2006). Two LRTs, M7-M8 and M8A-M8, were significant at the 0.01 and 0.05 significance level, respectively. Comparing M8 against the null model M8A is more robust than M1A versus M2A or M7 versus M8 (Swanson et al., Reference Swanson, Nielsen and Yang2003).
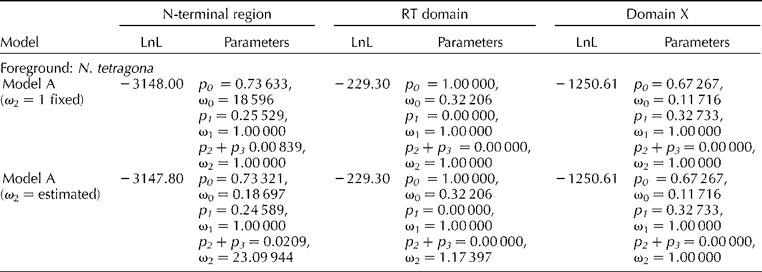
Similarly, branch-site model A (ω2 = 1 fixed) with branch-site model A (ω2 estimated) were compared. For N-terminal region, model A (ω2 estimated) for N. tetragona branch indicated that 2.09% of the sites are under selective pressure with ω2 = 23.099 (Table 1). Bayes Empirical Bayes (BEB) analysis showed three positive sites with probability value of 0.607, 0.527 and 0.582, respectively.
Table 1 Estimation of log-likelihood (LnL) values and parameters for each region of the matK gene using branch-site models implemented in PAML selecting N. tetragona as foreground branch.
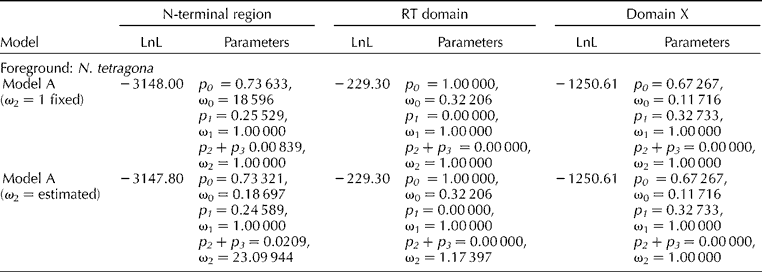
p, proportion of codon sites with ω; ω, dN/dS.
Selective pressure during colonization of N. tetragona
N. tetragona is well distributed globally, found throughout China, Japan, Finland and Russia; but its location in India is restricted to one particular population. Our previous study indicated that the Indian plant is closely related to plants found in China (Dkhar et al., Reference Dkhar, Kumaria, Rama Rao and Tandon2010). Molecular evolutionary analysis suggested that the plant taxon migrated from China, and the migratory processes involved might have brought about some changes at both the DNA and protein sequence levels. This adaptive response would have rendered slight advantage for the plant to survive in new habitat. Such adaptive changes at the DNA and protein sequence levels of rbcL gene have been reported in Schidea (Kapralov and Filatov, Reference Kapralov and Filatov2006) and Potamogeton (Iida et al., Reference Iida, Miyagi, Aoki, Ito, Kadono and Kosuge2009).
Implications for conservation of N. tetragona
Determining the genetic makeup of a particular plant species is of critical importance when planning a suitable conservation strategy. Here, we provided evidence for molecular adaptation of matK gene in N. tetragona, a critically rare and endangered plant of India. Such adaptive changes at the molecular level may have been associated with the adaptation of N. tetragona to varying ecological conditions. So, the threat to the existence of this plant taxon is primarily anthropogenic, brought about through large-scale cultivation at the vicinity of the location. Conservation of N. tetragona can be targeted at controlling such activities or the probable translocation to another site.
Acknowledgements
The authors acknowledge financial support from Department of Biotechnology, Ministry of Science and Technology, India (file no. BT/PR-7055/BCE/08/437/2006).