INTRODUCTION
In 2017, a small collection of drugs and treatments are used and recommended for visceral leishmaniasis (VL) (WHO, 2010; Aronson et al. Reference Aronson, Herwaldt, Libman, Pearson, Lopez-Velez, Weina, Carvalho, Ephros, Jeronimo and Magill2017) and even fewer that have proven effective in the treatment of cutaneous leishmaniasis (CL) (González et al. Reference González, Pinart, Reveiz and Alvar2008, Reference González, Pinart, Rengifo-Pardo, Macaya, Alvar and Tweed2009; Aronson et al. Reference Aronson, Herwaldt, Libman, Pearson, Lopez-Velez, Weina, Carvalho, Ephros, Jeronimo and Magill2017). Of these, and still widely in use, are the pentavalent antimonials (Pentostam has been in use since the 1940s; Goodwin, Reference Goodwin1995), amphotericin B, which was first used for leishmaniasis in 1960 (Sampaio et al. Reference Sampaio, Godoy, Paiva, Dillon and de Lacaz1960) and as a liposomal formulation in 1991 (Davidson et al. Reference Davidson, Scott, Maini, Bryceson and Croft1991), paromomycin first used for leishmaniasis in 1963 (Kellina et al. Reference Kellina, Iniakhina and Iastrebova1966), and miltefosine identified as anti-leishmanial in 1984 (Croft et al. Reference Croft, Neal, Pendergast and Chan1987) and registered for use for VL treatment in 2002 (Sundar et al. Reference Sundar, Singh, Rai, Prajapati, Singh, Ostyn, Boelaert, Dujardin and Chakravarty2012). The limitations of these drugs and treatments have been reviewed elsewhere (Croft, Olliaro, Reference Croft and Olliaro2011; Aronson et al. Reference Aronson, Herwaldt, Libman, Pearson, Lopez-Velez, Weina, Carvalho, Ephros, Jeronimo and Magill2017). It is worth noting that these drugs providing the standard treatments for VL and CL are mainly re-purposed. To help improve the drug R & D process it is important to ask why it is taking so long to identify purpose-designed anti-leishmanials.
Approaches to the design, discovery and development of anti-infectives have advanced considerably over the past two decades and several unique molecular and biochemical targets of parasites/microbes are the subject of other articles in this volume. For leishmaniasis advances in molecular biology and structural biology have similarly led the elaboration of validated targets and inhibitors (Gilbert, Reference Gilbert2013; Horn and Duraisingh, Reference Horn and Duraisingh2014), whilst high-throughput (HTS) and high content (HCS) have led to the identification of novel chemical series (Siqueira-Neto et al. Reference Siqueira-Neto, Moon, Jang, Yang, Lee, Moon, Chatelain, Genovesio, Cechetto and Freitas-Junior2012; Peña et al. Reference Peña, Manzano, Cantizani, Kessler, Alonso-Padilla, Bardera, Alvarez, Colmenarejo, Cotillo, Roquero, de Dios-Anton, Barroso, Rodriguez, Gray, Navarro, Kumar, Sherstnev, Drewry, Brown, Fiandor and Martin2015), as well as the identification of novel targets (Khare et al. Reference Khare, Nagle, Biggart, Lai, Liang, Davis, Barnes, Mathison, Myburgh, Gao, Gillespie, Liu, Tan, Stinson, Rivera, Ballard, Yeh, Groessl, Federe, Koh, Venable, Bursulaya, Shapiro, Mishra, Spraggon, Brock, Mottram, Buckner, Rao and Wen2016). A pragmatic use of screening and extension of chemical series with known anti-kinetoplastid activity has produced lead compounds, and novel chemical entities (NCEs), from oxaboroles, nitroimidazoles, aminopyrazoles; which are all promising candidates in the anti-leishmanial development pipeline (http://www.dndi.org).
The parts played by pharmacokinetics (PK) and medicinal chemistry have also lead to improved design of compounds able to target pathogens in infected tissues, for example in the CNS (Wring et al. Reference Wring, Gaukel, Nare, Jacobs, Beaudet, Bowling, Mercer, Bacchi, Yarlett, Randolph, Parham, Rewerts, Platner and Don2014) and the macrophage (Rajendran et al. Reference Rajendran, Knölker and Simons2010), tissues that are relevant to the distribution of anti-trypanosomal and anti-leishmanial agents. In addition, the past decade has seen the integration of PK, pharmacodynamics (PD) and physiology-based (PB) modelling, as illustrated in recent reviews on PK–PD analysis (Nielsen and Friberg, Reference Nielsen and Friberg2013) and PB–PK analysis (Edginton et al. Reference Edginton, Theil, Schmitt and Willmann2008), into drug design and the prediction of appropriate dosing of novel and current anti-infective drugs. The importance of PK–PD analysis has been well demonstrated for drug design for Mycobacterium tuberculosis (Davies and Nuermberger, Reference Davies and Nuermberger2008; Dartois, Reference Dartois2014), a pathogen that occupies a similar intracellular site as Leishmania. Another recent approach, using small molecules, has been to target host factors/receptors and nutrient sources, as considered for M. tuberculosis (Guler and Brombacher, Reference Guler and Brombacher2015; Zumla et al. Reference Zumla2015), and in a more limited way for leishmaniasis, for example with simvastatin (Parihar et al. Reference Parihar, Hartley, Hurdayal, Guler and Brombacher2016). Modulation of the host's immune response has been a long-term goal, with some small molecules showing activity through known targets, for example, imiquimod (Buates and Matlashewski, Reference Buates and Matlashewski1999) and tucaresol (Smith et al. Reference Smith, Yardley, Rhodes and Croft2000).
LEISHMANIA, PHYSIOLOGICAL BARRIERS AND DRUG DISTRIBUTION
There is nothing new about the concept of selectivity and drug distribution as the basis for the design, discovery and development of anti-infective drugs. Over the past century our work has been framed by the likes of Ehrlich (Reference Ehrlich1913), ‘…we may speak of magic bullets which aim exclusively at the dangerous intruding parasites, strangers to the organism, but do not touch the organism, itself and its cells…’. Alberts (Reference Alberts1985) also described the challenges of achieving drug selective toxicity, comparing them at three levels – whole-body distribution, biochemistry and cytology, while linking the physicochemical properties of compounds/drugs to pharmacodynamic effects.
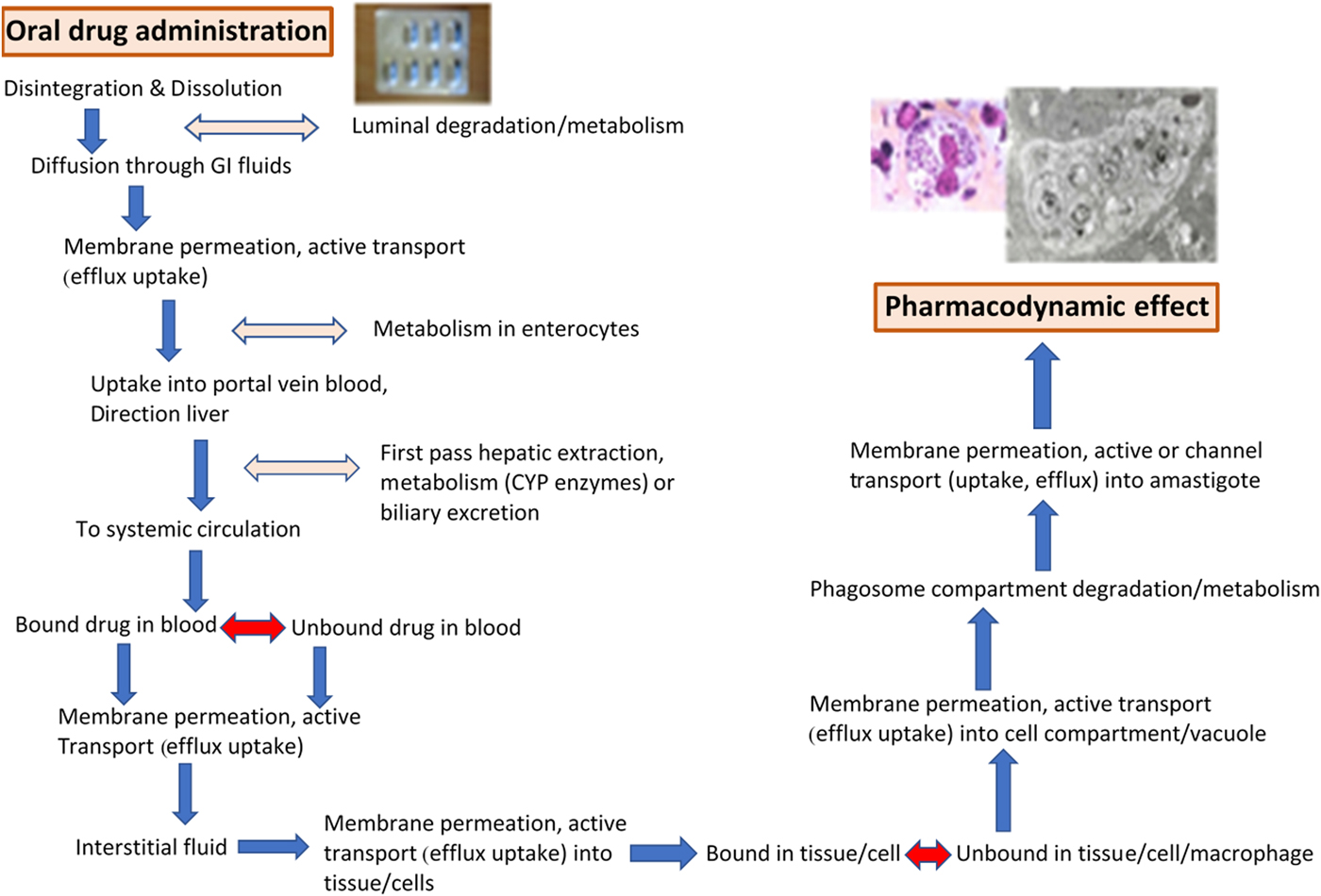
At the start of the drug discovery process it is essential to consider both: (i) the TPP (target product profile) of the drug needed (see, for example, http://www.dndi.org/diseases-projects/target-product-profiles) and (ii) the required distribution and PK of novel compounds to infected tissues. These considerations have to be integrated into the discovery and development of new treatments for both VL and CL. The design of oral drugs or topical formulations for the treatment of VL and/or CL has to account for the number of physiological barriers that the anti-leishmanial molecule meets before reaching the molecular target. Both the series of membrane barriers and the different protein-binding properties in plasma and tissues (Fig. 1), are challenges to be met before the novel compound reaches the macrophage host cell in which the Leishmania amastigote survives and multiplies (Fig. 2). For CL drugs, there are the additional barriers of extravasation and the interstitial fluid compartment, factors often not considered in this complex design. These challenges are not exclusive to those working on drug R & D for Leishmania; similar problems arise for intracellular bacteria, in particular Mycobacteria, and we can learn much from that research (Dartois, Reference Dartois2014).
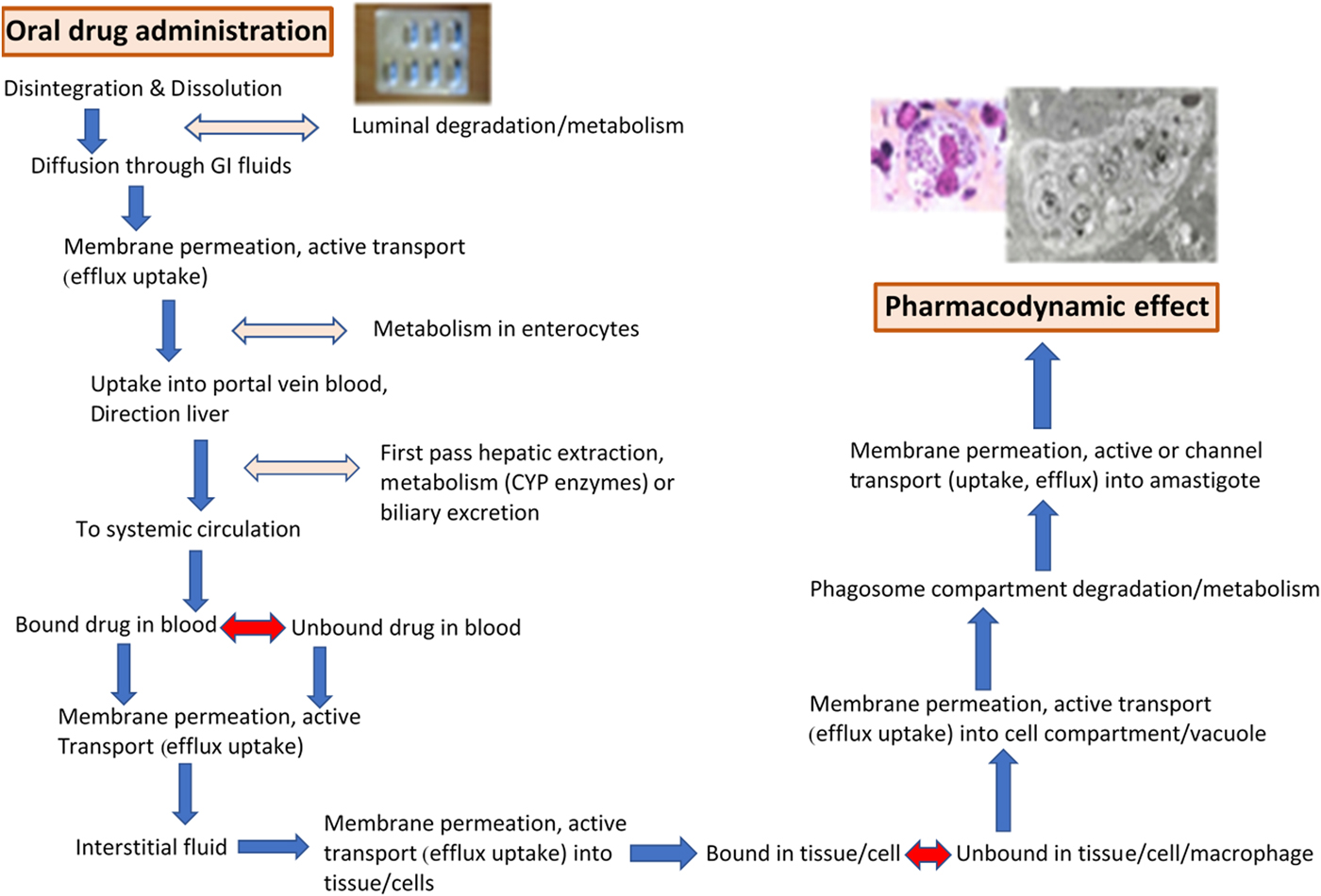
Fig. 1. The physiological barriers and pharmacokinetic requirements for an active compound in an oral drug (miltefosine capsules shown) to reach the Leishmania amastigote (adapted from Martinez, Amidon, 2002, Journal of Clinical Pharmacology, 620–643). Light and electron micrographs of L. donovani amastigotes.

Fig. 2. Leishmania donovani amastigotes in mouse liver Kupffer cell, showing blood vessel–host cell interface [bar + 1uM].
So, how can our knowledge of tissue and cellular distribution be exploited to improve drug design? How can our understanding of membrane barriers and protein binding improve our ability to ensure drugs reach the sites where they are needed? How can formulations and drug delivery be best exploited for this purpose?
SELECTIVITY AND PHARMACODYNAMICS
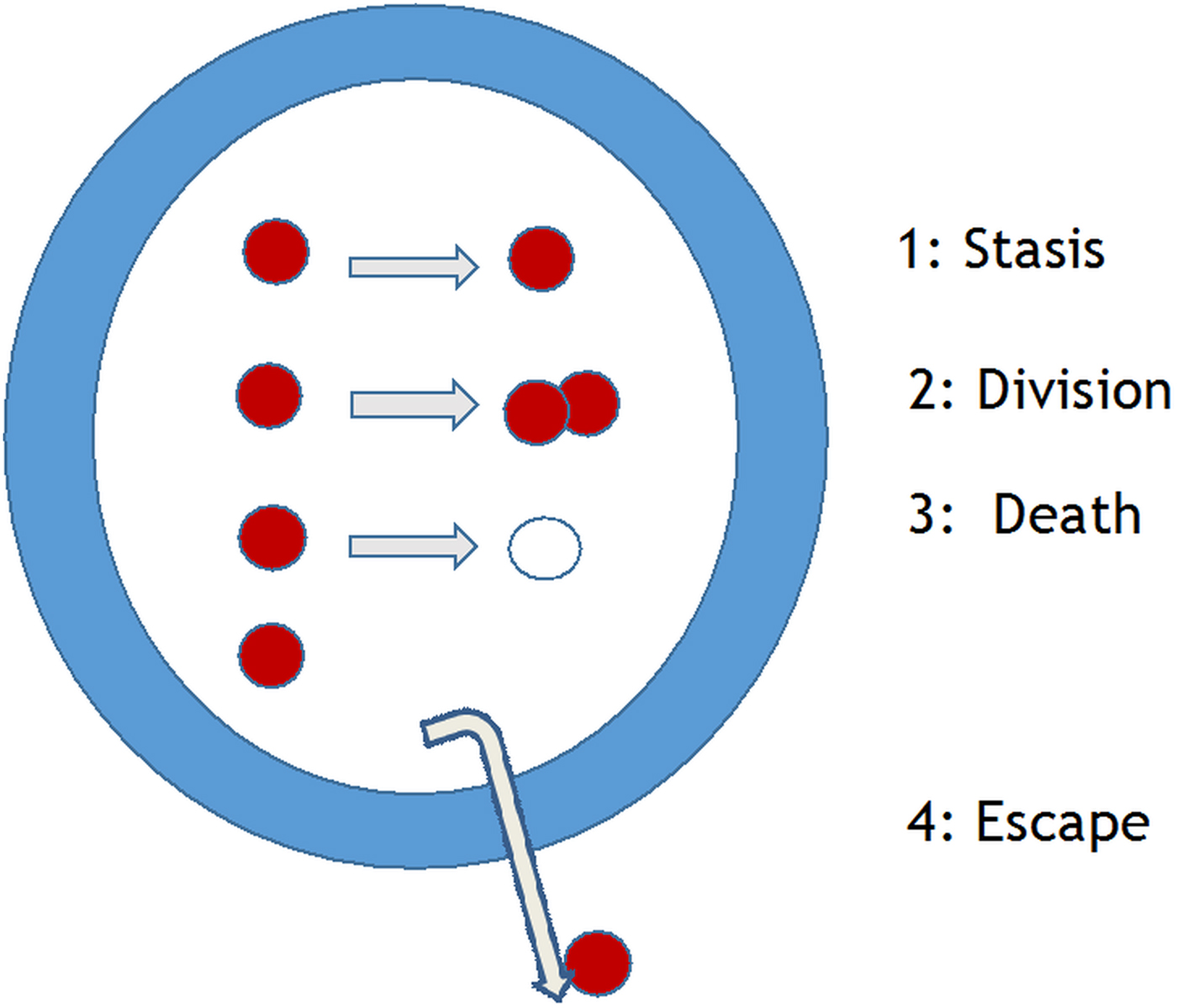
The initial identification of hit and lead compounds is normally determined in in vitro assays. Criteria for in vitro assays to measure killing of the intracellular Leishmania amastigote, were listed several decades ago (Croft, Reference Croft1986); this list needs to be updated to include: (i) the rate of kill, which has been shown to be important for other protozoa (Sanz et al. Reference Sanz, Crespo, De-Cózar, Ding, Llergo, Burrows, García-Bustos and Gamo2012), (ii) the type of macrophage used (see Seifert et al. Reference Seifert, Escobar and Croft2010 below), and (iii) a panel of recent clinical isolates, as there is known strain/species variation in drug sensitivity (Croft et al. Reference Croft, Sundar and Fairlamb2006). The standard phrase to introduce the Leishmania parasite in many reviews is ‘Leishmania survive and multiply in host macrophages’. Leaving aside immunological issues of macrophage activation (Kaye and Scott, Reference Kaye and Scott2011), measurements of drug or NCE effects should relate to potency against a dividing population of amastigotes in a defined macrophage population. However, there are four possible states for an amastigote in the macrophage (Fig. 3): stasis, division, death or escape. These issues, are also a major concern for M. tuberculosis researchers, but have only recently been investigated for Leishmania. In vitro studies have given some measures of rate of division through 3H-thymidine uptake (Sifontes and Croft, unpublished) and the bromodeoxyuridine analogue (EDU) (Tegazzini et al. Reference Tegazzini, Díaz, Aguilar, Peña, Presa, Yardley, Martin, Coteron, Croft and Cantizani2016), both methods indicating incorporation of these indicators of nucleic acid synthesis into about 50% of Leishmania donovani amastigotes in macrophage assays. In the steps from in vitro assay to rodent model and from rodent model to human, it is essential to know the ‘predictivity’ of the model. So, are in vitro assays with rapid rates of division more predictive of activity in the rodent model than those with slow rates of division? Some in vivo studies suggest that the answer is possibly not. Recent studies on Leishmania major and Leishmania mexicana amastigotes in mouse models have shown that: (i) L. mexicana amastigote division rate is slow (12 days) and sub-populations develop that are either semi-quiescent or fast-growing state (Kloehn et al. Reference Kloehn, Saunders, O'Callaghan, Dagley and McConville2015) and (ii) L. major amastigotes in lesions similarly show fast and slow replicating populations (Mandell and Beverley, Reference Mandell and Beverley2017). Similar questions must now be asked about L. donovani liver and spleen infections in mouse and hamster models to understand the relevance and predictivity of our current models. This underlines the frailty of the PD parameters currently applied in PK–PD analysis for Leishmania models. It would be a great benefit to Leishmania research to be able to transfect with reporter genes that reflect in vivo growth rates. In studies on Salmonella different bacteria cell division rates in different tissues were shown to correlate with different levels of killing by antibiotics (Claudi et al. Reference Claudi, Spröte, Chirkova, Personnic, Zankl, Schürmann, Schmidt and Bumann2014).
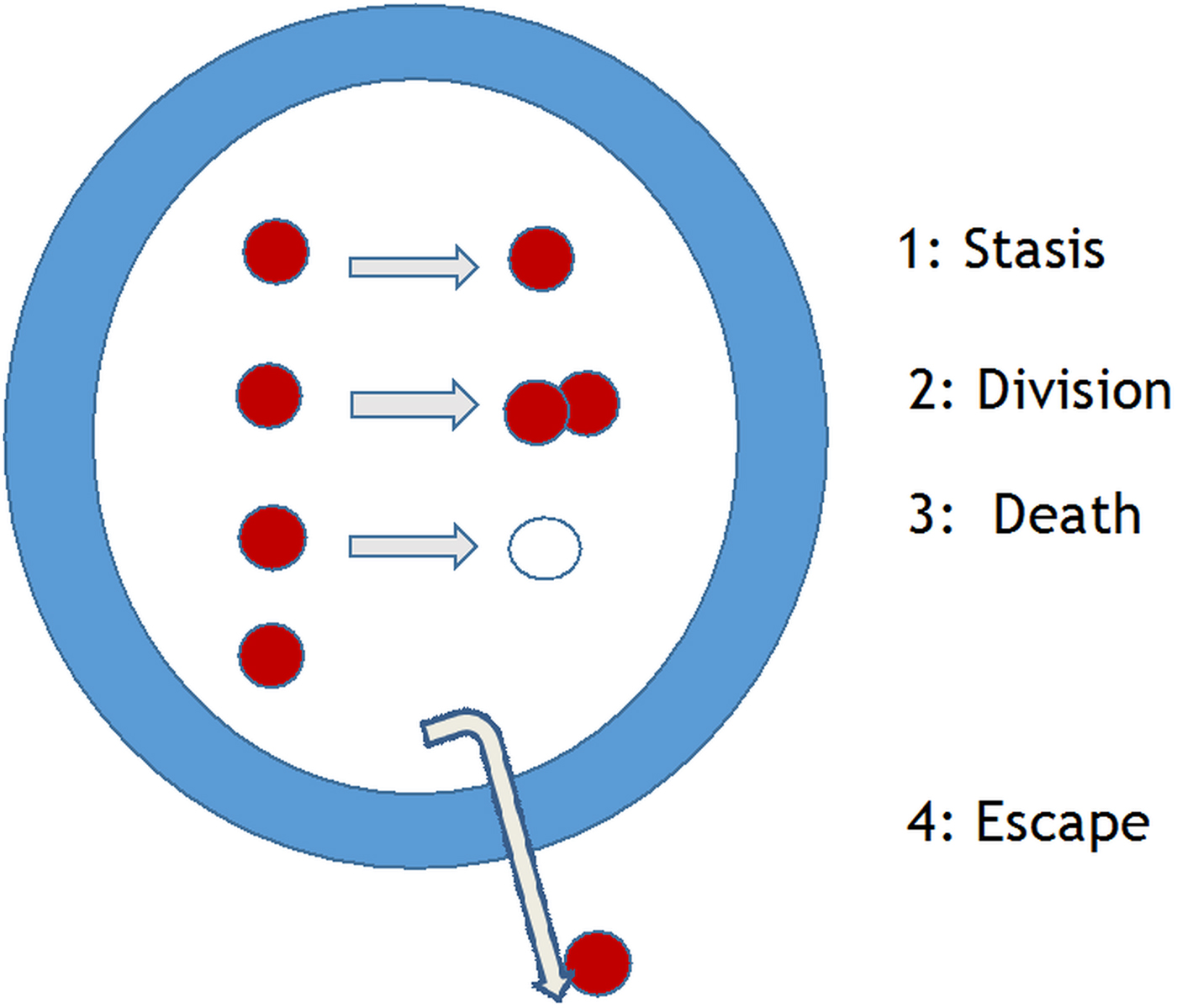
Fig. 3. Leishmania amastigotes – potential four fates of the intracellular amastigote in a macrophage [Courtesy of R. Diaz-Gonzalez and S. Croft].
In addition to the above concerns about ‘predictivity’, most amastigote–macrophage in vitro assays, the current workhorse of anti-leishmanial drug discovery, over the past three decades, have used either macrophage cell lines (e.g. THP-1 cells) or primary isolated rodent macrophages (Neal and Croft, Reference Neal and Croft1984; De Rycker et al. Reference De Rycker, Hallyburton, Thomas, Campbell, Wyllie, Joshi, Cameron, Gilbert, Wyatt, Frearson, Fairlamb and Gray2013). These have on the whole been useful in hit and lead identification. Whether there are more appropriate assays for further evaluation and lead identification has been questioned. Ex vivo models with splenic and lymph node tissue have been used in drug assays (Osorio et al. Reference Osorio, Travi, Renslo, Peniche and Melby2011; Peniche et al. Reference Peniche, Renslo, Melby and Travi2014) to try to more closely match host physiological conditions. Recently we have developed a medium perfusion/flow system, using interstitial flow rates, with L. major-infected macrophages. There is a shift in the dose–response curves of several standard anti-leishmanial drugs in flow vs static systems (O'Keeffe, Croft, unpublished), where EC50 points were similar but EC90 points were significantly lower in the flow model.
MACROPHAGES
The host macrophage is a central consideration in drug design and targeting in addition to its role in vitro drug assays, from monocytic cell lines (such as THP-1, J774 lines) used in HTS, to murine peritoneal or bone marrow macrophages used in evaluation and mechanistic studies. The role of the macrophage in the immune response, pathogenesis, and parasite invasion and survival in the phagosomal compartment has been well described elsewhere (Kaye and Scott, Reference Kaye and Scott2011). One ‘macrophage’ factor that is important in therapy and certainly in deriving PD parameters, is the heterogeneity of the macrophage cell population (Gordon et al. Reference Gordon, Plüddemann and Martinez Estrada2014) with different cell types having different functions and properties in different tissues. Recent in vitro studies with L. donovani in amastigote macrophage assays has shown that significant differences in amastigote drug susceptibility are macrophage-type dependent (Seifert et al. Reference Seifert, Escobar and Croft2010; Koniordou et al. Reference Koniordou, Patterson, Wyllie and Seifert2017). This is not just an issue for standardization of assays, it is also important in understanding differences in host cell drug accumulation and role of transporters, as well as drug metabolism, for example pentavalent antimonials are metabolized by macrophages (Frézard et al. Reference Frézard, Demicheli and Ribeiro2009) and their activity altered through macrophage activation (Murray et al. Reference Murray, Berman and Wright1988). The changes in macrophage metabolism with Leishmania infection could also impact drug activity and be different between macrophage populations, as has been shown for M. tuberculosis using RNA-seq analysis (Andreu et al. Reference Andreu, Phelan, de Sessions, Cliff, Clark and Hibberd2017). Connections between drug uptake to cell type, infection and activation status have long been defined for anti-bacterials (Carlier et al. Reference Carlier, Scorneaux, Zenebergh, Desnottes and Tulkens1990). With new sensitive methods to measure anti-leishmanial drug accumulation into immune cells (Kip et al. Reference Kip, Rosing, Hillebrand, Castro, Gomez, Schellens, Beijnen and Dorlo2015) further studies to define Leishmania–host cell–drug interactions are needed.
There is also opportunity to exploit the physicochemical properties of the phagolysosomal compartment, by manipulating conditions to improve drug activity, as exemplified by doxycline treatment of Coxiella infection (Maurin et al. Reference Maurin, Benoliel, Bongrand and Raoult1992). The relationship between the pH of the phagolysosomal vacuole and amastigote survival is well established and pH manipulation with basic drugs, such as chloroquine, leads to parasite death. We have recently shown that in combination with chloroquine the activity of paromomycin can be significantly enhanced against L. major and L. mexicana amastigotes in vitro in mouse peritoneal macrophages (Wijnant et al. Reference Wijnant, Van Bocxlaer, Yardley, Murdan and Croft2017a ). The acidic environment of the lysosomal compartment of cells has been studied in relation to the accumulation of charged basic molecules with dicationic molecules such as azithromycin having high accumulation levels (see Van Bambeke et al. Reference Van Bambeke, Barcia-Macay, Lemaire and Tulkens2006). The potential for concentration and trapping through protonation of novel molecules was exquisitely exploited by Rabinovitch et al. (Reference Rabinovitch, Zilberfarb and Ramazeilles1986) using amino acid esters to kill L. amazonensis amastigotes in mouse peritoneal macrophages. Regulation of the phagosome compartment occurs through a number of membrane enzymes and transporters, including the well-characterized vacuolar proton-ATPase (v-ATPase) involved in vacuole acidification and biogenesis (Vinet et al. Reference Vinet, Jananji, Turco, Fukuda and Descoteaux2011) and the cation transporter Nramp 1 (Jabado et al. Reference Jabado, Jankowski, Dougaparsad, Picard, Grinstein and Gros2000). Vacuolar enzyme and pH manipulation is being exploited in anti-cancer therapies, where the actions of known inhibitors synergize with standard drugs, with an impact on chemosensitization and reversal of chemoresistance (Swietach et al. Reference Swietach, Hulikova, Patiar, Vaughan-Jones and Harris2012). This is an area little studied in relation to Leishmania; one report has shown naloxonazine can upregulate V-ATPase in L. donovani-infected THP-1 cells (De Muylder et al. Reference Muylder, Vanhollebeke, Caljon, Wolfe, McKerrow and Dujardin2016). Outside the phagolysomal vacuole, the metabolic changes seen in macrophage biology and the numerous pathways altered by Leishmania infection, from iron to cholesterol and beyond has been recently reviewed (Duque and Descoteaux, Reference Duque and Descoteaux2015).
Finally, the macrophage cell surface has several defined receptors that have been used in drug targeting to these host cells. 25 years ago Nègre et al. (Reference Nègre, Chance, Hanboula, Monsigny, Roche, Mayer and Hommel1992), showed that allopurinol riboside linked to mannosylated – poly-L-lysine as the carrier molecule to target the mannose receptor, improved drug activity 50-fold in an in vitro macrophage model. There have been several other publications describing tagged liposomes or nanoparticles in experimental models to target and increase drug accumulation by infected macrophages. However, given the added complexity of the synthesis, costs, stability and PK, it is unlikely that this approach will lead to new treatments for leishmaniasis.
PK AND PREDICTIVE MODELS
As part of the effort to reduce the attrition rates that occur at each stage of the drug R & D process – from in vitro to animal models, from animal models to clinical candidate, and then in clinical trials – major efforts have been made to develop more predictive models to advance optimized novel compounds at each stage (for pharmacodynamics see above) and their behaviour in animal models and humans, i.e. PK. PKs normally encompass the properties of a novel compound/drug of absorption, distribution, metabolism and excretion. The anti-infective drug researcher also needs to ensure appropriate distribution of the compound to the infected tissue in the host, as well as retention of the compound within the infected tissue/cells for a period to give sufficient exposure to significantly reduce the parasite load. Over the past two decades there has been an increasing focus on the interrelationship between PDs and PKs, including tissues other than just plasma in the analysis. The PK–PD paradigms were first outlined for anti-bacterials (Craig, Reference Craig1998) with an initial focus on how this information, which often focuses on defining drug activities in terms of either concentration dependent rate of killing or time-dependent rate of killing, can be used to help determine effective dose regimens. For anti-leishmanials PK–PD analysis has helped to re-define miltefosine dosing in children for both VL and CL (Dorlo et al. Reference Dorlo, Balasegaram, Beijnen and de Vries2012; Castro et al. Reference Castro, Gomez, Kip, Cossio, Ortiz, Navas, Dorlo and Saravia2017). However, for most anti-leishmanial drugs we have little data on their time-dependent or concentration-dependent activities, nor how these data can be used to optimize dose regimes. Analytical and mathematical tools are available to simulate PK/PD relationships of anti-leishmanials when there is sufficient and appropriate data. One such system has been developed for anti-malarial pre-clinical development, which has helped to identify the properties of compounds important for clinical performance and, importantly, partner drugs for combination therapy (Patel et al. Reference Patel, Simpson, Batty, Zaloumis and Kirkpatrick2015; Aljayyoussi et al. Reference Aljayyoussi, Kay, Ward and Biagini2016).
This analysis of the PK–PD relationship has also become central to drug development. A Pfizer team analysed the role of fundamental PK–PD as part of a process to improve the survival rate of compounds in clinical trials, i.e. what predictive indicators are of most importance to reduce attrition. They defined ‘three pillars of survival’ for a novel compound as: (i) exposure at the target site of action over a desired period of time; (ii) binding to the pharmacological target as expected for its mode of action; and (iii) expression of pharmacological activity commensurate with the demonstrated target exposure and target binding (Morgan et al. Reference Morgan, Van Der Graaf, Arrowsmith, Feltner, Drummond, Wegner and Street2012). An important element of PK–PD analysis is that in vivo only the free (i.e. not bound to protein) drug concentration determines activity (Smith et al. Reference Smith, Di and Kerns2010); hence for Leishmania knowledge of drug concentration in the phagosome vacuole is of importance but so far undetermined. For leishmaniasis, we have limited retrospective data on miltefosine, as summarized by Dorlo et al. (Reference Dorlo, Balasegaram, Beijnen and de Vries2012). More research has been reported on the liposomal formulation of amphotericin B, the unilamellar liposome AmBisome ™. We have shown that in a BALB/c mouse model (with data from liver, spleen and plasma) that following iv dosing of AmBisome parasites in the liver are killed quicker and more effectively than those in the spleen (Voak et al. Reference Voak, Harris, Qaiser, Croft and Seifert2017). This can be explained by more extensive drug accumulation by the liver than spleen and the different drug kinetics between the two organs, although drug accumulation in these target organs was higher during the early stages of infection than later stages. Earlier studies with AmBisome in mice showed lower amphotericin B accumulation in infected than uninfected mice and also that the formulation was less effective in the later stage than an early stage infection (Mullen et al. Reference Mullen, Baillie and Carter1998; Gershkovich et al. Reference Gershkovich, Wasan, Sivak, Li, Zhu, Werbovetz, Tidwell, Clement, Thornton and Wasan2010). The effectiveness of drugs in different tissues and distribution of drug in the tissues needs to be considered alongside the changes that occur in liver and spleen structure and function during early and late stages on infection (Yurdakul et al. Reference Yurdakul, Dalton, Beattie, Brown, Erguven, Maroof and Kaye2011; Kaye and Beattie, Reference Kaye and Beattie2016). Recent studies on M. tuberculosis drugs to study their spatial distribution in lung lesions used MALDI mass spectrometry imaging to reveal which TB drugs penetrated the lesions (Prideaux et al. Reference Prideaux, Via, Zimmerman, Eum, Sarathy, O'Brien, Chen, Kaya, Weiner, Chen, Song, Lee, Shim, Cho, Kim, Cho, Olivier, Barry and Dartois2015); this is an approach which should be applied to help us understand the tissue distribution of anti-leishmanials.
The use of transfected Leishmania parasites with either fluorescent or bioluminescent properties have been used in studies on infection, pathology and chemotherapy over the past decade. But in terms of analysing key properties of anti-leishmanial drug action, these methods are only now starting to be fully exploited, in particular in rodent models of several species causing CL where drug efficacy and relapse have been measured (Coelho et al. Reference Coelho, Oliveira, Espada, Reimão, Trinconi and Uliana2016; Caridha et al. Reference Caridha, Parriot, Hudson, Lang, Ngundam, Leed, Sena, Harris, O'Neil, Sciotti, Read, Lecoeur, Hickman and Grogl2017). However, with good signal and high-resolution imaging it will be important to improve the methodologies to measure: (i) dose response effect, (ii) in vivo rate of kill and (iii) any differences in drug efficacy between sites of infection.
Drug combinations, which are presently for leishmaniasis really co-administrations, have proved to be clinically advantageous in the treatment of VL (Sundar et al. Reference Sundar, Sinha, Rai, Verma, Nawin, Alam, Chakravarty, Vaillant, Verma, Pandey, Kumari, Lal, Arora, Sharma, Ellis, Strub-Wourgaft, Balasegaram, Olliaro, Das and Modabber2011). Current combinations of standard anti-leishmanial drugs are based solely on doses used in monotherapies. As novel oral compounds are developed over the next 5 years and genuine combinations are discussed, then drug combinations based upon knowledge of PK–PD components of partner drugs and their interactions will be needed, as shown for anti-malarials (Hastings et al. Reference Hastings, Hodel and Kay2016). We also know that major challenges for treatment are VL–HIV co-infections (Van Griensven et al. Reference Van Griensven, Zijlstra and Hailu2014). There have been some in vitro studies on efficacy of combined anti-retroviral/anti-leishmanial drug interactions (Costa et al. Reference Costa, Machado, Cavadas and do Céu Sousa2016) and indication of anti-leishmanial activity of HIV-protease inhibitors (Van Griensven et al. Reference Van Griensven, Diro, Lopez-Velez, Boelaert, Lynen, Zijlstra, Dujardin and Hailu2013). However, there have been few studies where a rational approach to fully understand interactions between anti-leishmanial and anti-retroviral drugs and how this knowledge could be used to design more effective treatments. In contrast to tuberculosis and malaria where drug-drug interactions have been well characterized (see University of Liverpool, UK website http://www.hiv-druginteractions.org) there is limited analysis on anti-leishmanial drugs.
The collection of more PD data (from HCS screens) and PK data with the need to include specific compartments for tissues infected and uninfected, plus the overriding need to integrate all to inform dosing of the novel drug in humans, has re-focused need for the application of computational, modelling and systems biology (Van der Greef and McBurney, Reference Van der Greef and McBurney2005; Zhao and Iyengar, Reference Zhao and Iyengar2012). Recent mathematical model of anti-malarials has focused on simulations of PK/PD to predict clinical activity of new compounds (Aljayyoussi et al. Reference Aljayyoussi, Kay, Ward and Biagini2016). For leishmaniasis, a disease caused by a parasite that survives, multiplies and subverts the immune responses of the host macrophage (Kaye and Scott, Reference Kaye and Scott2011), the elements of immunity and immunopathology also have to be built into any predictive model. A computational Petri net model of L. donovani infection and granulomas in mouse liver (Albergante et al. Reference Albergante, Timmis, Beattie and Kaye2013; Moore et al. Reference Moore, Moyo, Beattie, Andrews, Timmis and Kaye2013) illustrates another approach to disease simulation, an approach that is being further exploited to understand the relationship between the immune response and the activity and the PKs of anti-leishmanial drugs (http://www.crackit.org.uk/multiscale-model-minimise-animal-usage-leishmaniasis-drug-development and http://www.leishsim.org) .
CL, SKIN AND TOPICAL FORMULATIONS
Considering the variety of clinical manifestations and the impact of CL, there is a notable absence of drugs and treatments that are clinically effective (González et al. Reference González, Pinart, Reveiz and Alvar2008, Reference González, Pinart, Reveiz and Alvar2009); this is an area of research neglect. This does not only apply to the classical forms of CL; there is also a need for improved treatments offering shorter courses, and less toxic drugs for post kala-azar dermal leishmaniasis (PKDL), as cases are infective to sandflies (Hirve et al. Reference Hirve, Boelaert, Matlashewski, Mondal, Arana, Kroeger and Olliaro2016) and PKDL patients act as a human reservoir for transmission and as such are a threat to elimination and control programmes.
Most research on CL has been focussed on immunological responses to CL in mice and humans (Kaye and Scott, Reference Kaye and Scott2011; Scott and Novais, Reference Scott and Novais2016), leading to paradigms on T-cell responses, which in conjunction with developments in knowledge on skin immunity and inflammation (see Pasparakis et al. Reference Pasparakis, Haase and Nestle2014) has provided some new understanding of the pathogenesis of CL. There have been several studies over the past decade showing how this understanding of skin immune response can be exploited for treatment, with a good example being the use of imiquimod in mice and humans (Buates and Matlashewski, Reference Buates and Matlashewski1999; Miranda-Verastegui et al. Reference Miranda-Verastegui, Tulliano, Gyorkos, Calderon, Rahme, Ward, Cruz, Llanos-Cuentas and Matlashewski2009). More recently, other approaches to treatment have resulted from long-term human CL research in South America. Novais et al. (Reference Novais, Carvalho, Clark, Carvalho, Beiting, Brodsky, Carvalho and Scott2017) showed that NLRP3 inflammasome is activated by CD8+ T cell-mediated cytotoxicity and drives disease progression. This led to experimental studies in mice using a number of small molecule inhibitors of the inflammasome, for example, MCC950 and the diabetes drug glyburide. They showed that treatment with compounds that inhibit NLRP3 inflammasome activation, MCC950 or glyburide, failed to develop the severe disease seen in untreated mice.
Apart from the immunotherapy approaches, the design and delivery of small molecules after oral administration to the skin has to be considered in the context of vascularization (drug gradient and distance between blood vessel and site), an interstitial fluid compartment, the impact of inflammation on drug accumulation and extra-vasation, blood flow rate (slow) and local oxygen tension (low). Improved understanding of PK of anti-leishmanial drugs in the skin has come from clinical studies where Dorlo and colleagues provided data on both PK and PD in human CL (L. major) patients treated with miltefosine (Dorlo et al. Reference Dorlo, van Thiel, Huitema, Keizer, de Vries, Beijnen and de Vries2008) although establishment of the full relationship between exposure and response has yet to be determined. We are now also beginning to understand how local inflammation at the CL site of infection can lead to specific accumulation of some drugs, for example liposomal amphotericin B, to improve cure (Wijnant et al. Reference Wijnant, Van Bocxlaer, Yardley, Harris, Murdan and Croft2017b ).
A critical decision in the development pathway for CL treatment is whether to choose systemic or topical administration. Topical formulations have been used for the treatment of CL since the 1920s when stibosan (an early pentavalent antimonial) ointment was used to treat ‘oriental sore’. We have come a long way since 1935 when the use of an ointment consisting of ‘1 part pulverized vegetable charcoal and nine parts of concentrated sulphuric acid’ was described. However, the full exploitation of pharmaceutics and knowledge of skin, from utilization of knowledge of skin physiology and PB–PK models, alteration of vasculature, role of protein binding and other factors (Jepps et al. Reference Jepps, Dancik, Anissimov and Roberts2013), including lymphatic flow to deeper layers (Dancik et al. Reference Dancik, Anissimov, Jepps and Roberts2012), are only now being applied to the development of new treatments for CL. The renaissance in the topical approach was led by El-On et al. (Reference El-On, Jacobs, Witztum and Greenblatt1984) with paromomycin, using an irritant and transdermal enhancing agent (methyl benzethonium chloride) to increase drug permeation by pore formation, the basis for the product Leishcutan® (Teva, Israel). Another formulation of topical paromomycin, containing 15% paromomycin–0·5% gentamicin and several excipients (called WR 279,396) with known absorption and skin PK (Ravis et al. Reference Ravis, Llanos-Cuentas, Sosa, Kreishman-Deitrick, Kopydlowski, Nielsen, Smith, Smith, Ransom, Lin and Grogl2013) when applied with an occlusion, has successfully completed phase three trials (Ben Salah et al. Reference Ben Salah, Ben Messaoud, Guedri, Zaatour, Ben Alaya, Bettaieb, Gharbi, Belhadj Hamida, Boukthir, Chlif, Abdelhamid, El Ahmadi, Louzir, Mokni, Morizot, Buffet, Smith, Kopydlowski, Kreishman-Deitrick, Smith, Nielsen, Ullman, Norwood, Thorne, McCarthy, Adams, Rice, Tang, Berman, Ransom, Magill and Grogl2013).
Focus on potency, permeation, and distribution (Jepps et al. Reference Jepps, Dancik, Anissimov and Roberts2013) is important for both formulation design and the selection of appropriate compounds with both high potency and, through their chemical properties, dermal distribution. As part of our strategy, we : (i) identify novel compounds that work against a panel of clinical isolates of the 15 species, as Leishmania species that cause CL vary significantly in their drug susceptibility (Escobar et al. Reference Escobar, Matu, Marques and Croft2002; Croft et al. Reference Croft, Sundar and Fairlamb2006); (ii) select active compounds with appropriate medicinal chemistry, toxicity and ADME (absorption, distribution, metabolism, excretion) properties; (iii) test in mouse models of infection (oral and systemic administration); (iv) optimize for compound structure and formulations in relation to skin distribution; (v) decide whether topical administration is appropriate and further optimize the formulations using both mouse and human skin in permeation studies; and (vi) ensure that treatment is effective against early-stage infections with intact skin (prior to ulceration) as the aim is to develop a treatment effective before the patient has developed into a large disfiguring ulcer.
When considering the PK of drugs for CL, it is important to remember that the Leishmania are in macrophages in the skin dermis, and that the infection is not superficial like many bacterial or fungal infections. Within the dermis at the site of infection, there is well-characterized inflammation and granuloma development (Scott and Novais, Reference Scott and Novais2017) and amastigotes in macrophages. This is found in both the nodule that precedes ulceration or later in the dermal rim around the ulcer. The aim for a systemic formulation is penetration from the vasculature via the interstitial fluid and distribution to the inflammatory site of infection. However, for a topical formulation the aims are permeation of the skin barriers and then distribution to the site of infection. In both cases exposure following distribution and residence of the compound at the site of infection has to be optimized. Some of the practices of pharmaceutical scientists working on skin for cosmetics and other purposes have been adopted for CL studies over the past decade. Using methodologies like the Franz cell, it is possible to measure the rate of diffusion of anti-leishmanial drugs across skin (of animal models and humans) alone and in different formulations, as shown for buparvaquone where the most effective topical formulation in vivo proved to be the one that crossed the skin most slowly in the Franz cell model (Garnier et al. Reference Garnier, Mäntylä, Järvinen, Lawrence, Brown and Croft2007a , Reference Garnier, Mäntylä, Järvinen, Lawrence, Brown and Croft b ). Recently this work has been extended to include Leishmania-infected skin. Permeation markers, for example caffeine and ibuprofen, as well as some standard anti-leishmanial drugs have been shown to have different in vitro permeation properties through normal mouse skin, compared with mouse skin removed from a nodule of infection (Van Boxclaer et al. Reference Van Bocxlaer, Yardley, Murdan and Croft2016a ). Drugs permeate significantly faster through skin taken from the site of infection, possibly due to oedema and the different immunological profile at this site of inflammation. As more extensive exposure in the dermis is critical to formulation design, the permeation properties of formulation excipients alone and together need to be explored. A re-examination of topical formulations of the anti-leishmanial drug miltefosine, using in vitro and in vivo models already mentioned, and a range of formulations in which the partition of miltefosine was characterized, was unable to identify a formulation with good permeation and efficacy (Van Bocxlaer et al. Reference Van Bocxlaer, Yardley, Murdan and Croft2016b ).
CONCLUSIONS FOR LEISHMANIASIS DRUG R & D
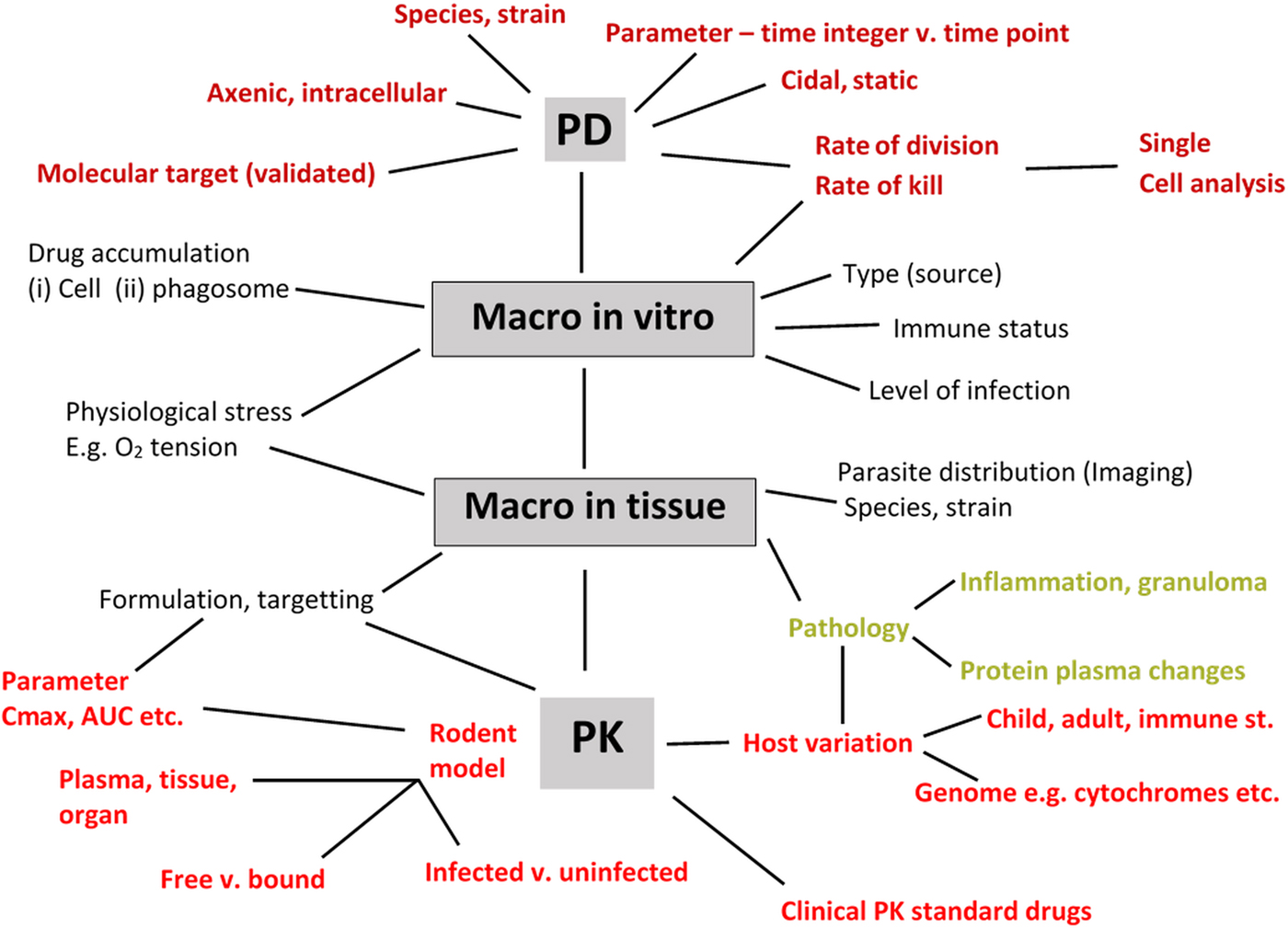
There have been several reviews that represent the drug R & D process as a linear diagram from discovery to clinical trial. However, drug R & D is a multi-disciplinary iterative process with many decision points, and the involvement of several teams across disciplines (Baxter et al. (Reference Baxter, Horn, Gal-Ed, Zonno, O'Leary, Terry and Terry2013). The parasitologist has key roles within this complex picture and an awareness of the comprehensive list of detailed information that he/she should aim to provide as part of a drug research team – ranging from work on enzyme targets to PK is needed. Although the concept of the ‘minimum information about a bioactive entity (MIABE)’ (Orchard et al. Reference Orchard, Al-Lazikani, Bryant, Clark, Calder, Dix, Engkvist, Forster, Gaulton, Gilson, Glen, Grigorov, Hammond-Kosack, Harland, Hopkins, Larminie, Lynch, Mann, Murray-Rust, Lo Piparo, Southan, Steinbeck, Wishart, Henning Hermjakob, Overington and Thornton2011) was established to provide guidance for what and how results should be reported, their review also provides a fundamental list of research information that needs to be gleaned from studies. In the specific area of leishmaniasis and Leishmania, where there are a large variety of assays and models involving different species, strains and stages of the Leishmania parasite, different host cells and different mammalian hosts, it is hardly surprising that there can be significant differences in data obtained between laboratories resulting in reports of irreproducibility of compound activities. In addition to basic precepts, such inclusion of controls, Fig. 4 is an attempt to summarize the main PD and PK-related factors that must be considered when collecting data during drug discovery and early pre-clinical studies for a novel anti-leishmanial compound. Although there are benefits for standardization, a process necessary when determining drug sensitivity of clinical isolates (Hendrickx et al. Reference Hendrickx, Guerin, Caljon, Croft and Maes2017), it is hardly feasible in the drug R & D process. But it is feasible for all those concerned to provide the levels of information sought and provided (Orchard et al. Reference Orchard, Al-Lazikani, Bryant, Clark, Calder, Dix, Engkvist, Forster, Gaulton, Gilson, Glen, Grigorov, Hammond-Kosack, Harland, Hopkins, Larminie, Lynch, Mann, Murray-Rust, Lo Piparo, Southan, Steinbeck, Wishart, Henning Hermjakob, Overington and Thornton2011) so that data can be interpreted by all those interested in playing a role in the development of the next drug and treatment for leishmaniasis.
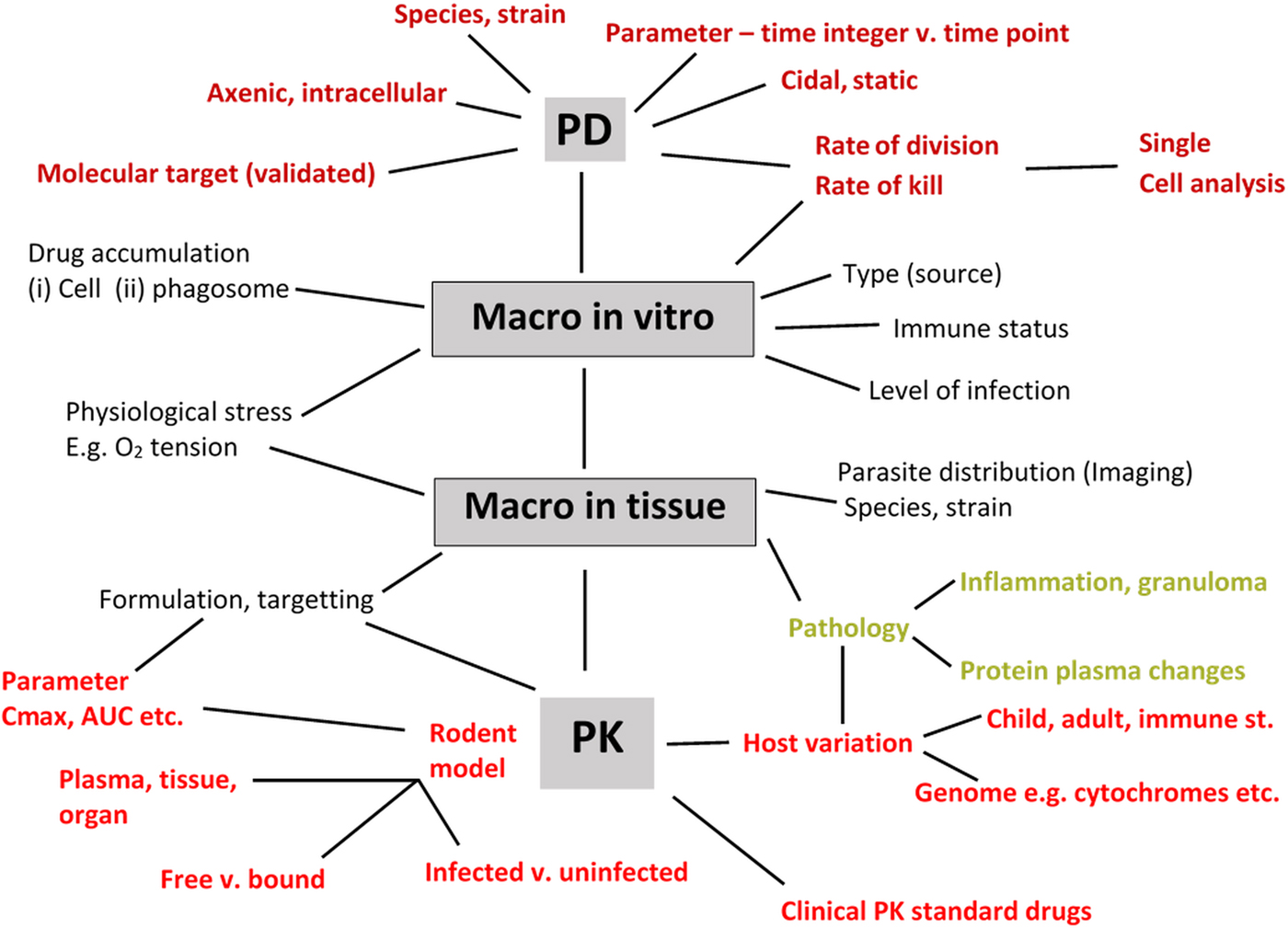
Fig. 4. Information to collect to support discovery and early pre-clinical development of a novel anti-leishmanial compound. PD – pharmacodynamics (axenic, intracellular amastigotes); PK – pharmacokinetics (C max: maximum concentration in plasma; AUC: Area-under-the curve as measure of drug exposure; macro – macrophage; free vs bound – protein-binding properties of compound.
ACKNOWLEDGEMENTS
The author is grateful to innumerable colleagues, partners, collaborators and mentors over the past decades whose advice, comments and input have contributed to this review.
FINANCIAL SUPPORT
The author has been supported recently by UK Medical Research Council (MRC), GSK Open Lab Foundation, NC3Rs CRACK IT, EU FP7 programme, DNDi Geneva and BBSRC for research on leishmaniasis.