


Introduction
Oil palm represents the single largest source of vegetable oils worldwide, producing 45.8 million tons of oil in 2010 or constituting 26.7% of the world's production of edible oils (FAOSTAT, 2010). This crop has rapidly expanded in the tropical countries of Asia, Africa and the Americas. Although the oil palm Elaeis guineensis Jacq. is native to Africa, the world's largest palm oil producers are Malaysia and Indonesia (Corley and Tinker, Reference Corley and Tinker2003).
Breeding is regarded as a practice that results in the reduction of genetic diversity. This reduction may change the future adaptability and breeding progress of the crop. Therefore, it is necessary to preserve the genetic variation that underlies the traits required for the sustainable development of oil palm cultivation. Crop wild relatives are particularly useful as a source of genes that confer resistance to various types of stress (Kuroda et al., Reference Kuroda, Tomooka, Kapa, Wanigadea and Vaughan2009). This has encouraged oil palm breeding programmes in Colombia to conduct exploratory studies on oil palm-derived materials in Angola and Cameroon for the establishment of ex situ genetic collections (Rey et al., Reference Rey, Gómez, Ayala, Delgado and Rocha2004; Arias et al., Reference Arias, Montoya and Romero2013a, Reference Arias, González, Prada, Restrepo and Romerob). The large number of oil palm accesions contained within these collections presented challenges for their conservation, evaluation, identification and usage. To promote the use of genetic resources, several authors have recommended the development of core collections. A core collection consists of a limited number of accessions that are selected to encompass the maximum genetic diversity and represent the genetic spectrum contained within the entire collection (Brown, Reference Brown1989). A core collection gives researchers more favourable access to the gene pool and can serve as a reference point for research studies, which, in turn, can provide an overview of the characteristics of the entire collection.
Molecular marker techniques have been used in applications such as the development of collection sampling and gap identification strategies, the planning of future collections, the management and conservation of germplasm banks, the identification of duplicates and genetic contamination, and the determination of genetic change associated with breeding (Hodgkin et al., Reference Hodgkin, Roviglioni, de Vicente and Dudnik2001). Molecular markers have been used for the development of core collections of both wild and cultivated plant species (Sangiri et al., Reference Sangiri, Kaga, Tomooka, Vaughan and Srinives2007; Hao et al., Reference Hao, Dong, Wang, You, Zhang, Ge, Jia and Zhang2008; Le Cunff et al., Reference Le Cunff, Fournier-Level, Laucou, Vezzulli, Lacombe, Adam-Blondon, Boursiquot and This2008; Kuroda et al., Reference Kuroda, Tomooka, Kapa, Wanigadea and Vaughan2009; Zhang et al., Reference Zhang, Zhang, Wang, Sun, Qi, Li, Wei, Han, Qiu, Tang and Li2011). These markers represent a powerful tool for germplasm characterization (Kalia et al., Reference Kalia, Rai, Kalia, Singh and Dhawan2011). Simple sequence repeats (SSRs) have been widely used to determine the genetic diversity of core collections of Elaeis from germplasm banks worldwide (Billotte et al., Reference Billotte, Risterucci, Barcelos, Noyer, Amblard and Baurens2001; Montoya et al., Reference Montoya, Arias, Rey and Rocha2005; Bakoumé, Reference Bakoumé, Wickneswari, Rajanaidu, Kushairi, Amblard and Billotte2007; Singh et al., Reference Singh, Mohd, Ting, Rosli, Tan, Leslie, Ithnin and Cheah2008; Cochard et al., Reference Cochard, Adon, Rekima, Billotte, Desmier de Chenon, Koutou, Nouy, Omoré, Purba, Glazsmann and Noyer2009; Arias et al., Reference Arias, Montoya and Romero2010; Billotte et al., Reference Billotte, Jourjon, Marseillac, Berger, Flori, Asmady, Adon, Singh, Nouy, Potier, Cheah, Rohde, Ritter, Courtois, Charrier and Mangin2010). Understanding of the genetic diversity and distribution of plants is essential for their preservation and usage. The objectives of this study were to use the molecular information generated by 29 SSR markers to determine the genetic diversity of oil palm accessions from Indonesia, Angola and Cameroon, and to select the entries that would constitute a core collection that ensured maximum allelic diversity.
Materials and methods
Plant material
The plant material used in this study is part of the ex situ collection of Cenipalma that is located in the Palmar de la Vizcaína experimental field (Barrancabermeja, Santander, Colombia). A total of 766 oil palm samples were analysed, of which 455 were collected from five geographical regions of the Republic of Angola, and 311 were collected from six geographical regions of the Cameroons (Rey et al., Reference Rey, Gómez, Ayala, Delgado and Rocha2004; Arias et al., Reference Arias, Montoya and Romero2013a, Reference Arias, González, Prada, Restrepo and Romerob). In addition, 22 samples of oil palm Deli dura collected from Indonesia were analysed. The seeds of these materials were acquired from the Bogor Botanical Gardens.
DNA extraction and SSR amplification
For the analysis, young palm leaflets were used. Each leaflet sample was macerated in liquid nitrogen, and DNA was isolated using a Qiagen extraction kit (reference no. 69106), following the manufacturer's protocol. The quality of the extracted DNA was evaluated by spectrophotometry and gel electrophoresis on a 0.8% agarose. DNA extracted from each sample was diluted to a concentration of 5 ng/μl for use in subsequent amplification reactions. A total of 29 microsatellite markers were used, and according to the linkage disequilibrium test, they were located at independent loci and mapped to 14 linkage clusters (Billotte et al., Reference Billotte, Marseillac, Risterucci, Adon, Brottier, Baurens, Singh, Herrán, Asmady, Billot, Amblard, Durand-Gasselin, Courtois, Asmono, Cheah, Rohde, Ritter and Charrier2005). The amplification conditions used for each marker have been described previously by Billotte et al. (Reference Billotte, Risterucci, Barcelos, Noyer, Amblard and Baurens2001) and Singh et al. (Reference Singh, Mohd, Ting, Rosli, Tan, Leslie, Ithnin and Cheah2008). The amplification products were resolved by electrophoresis on a 6% denaturing polyacrylamide gel containing 5 M urea and stained with silver nitrate, according to the protocols described by Qu et al. (Reference Qu, Li, Wu and Yang2005). Alleles were identified using a 10 base-pair (bp) molecular marker with fragment sizes ranging from 10 to 330 bp.
Data analysis
Genetic differentiation and diversity
Allele frequencies, total number of alleles, average polymorphic information content and allelic richness (R a) were calculated for each SSR locus, and evaluated based on the algorithms included in the (FSTAT software, http://www2.unil.ch/popgen/softwares/fstat.htm/; Goudet, Reference Goudet2002). Probability of identity, observed heterozygosity (H o) and expected heterozygosity (H e) were estimated for each locus using the (GIMLET software, http://pbil.univlyon1.fr/software/Gimlet/gimlet.htm/; Gimlet, Reference Gimlet2002). The number of private alleles identified for each geographical origin was calculated using the (GENALEX software, http://biology.anu.edu.au/GenALEx/; Peakall and Smouse, Reference Peakall and Smouse2006). Genetic differentiation among the geographical origins was determined based on Nei's (Reference Nei1987) coefficient of genetic diversity and differentiation using the (FSTAT software, http://www2.unil.ch/popgen/softwares/fstat.htm/; Goudet, Reference Goudet2002).
Establishment of the core collection
Here, the term ‘accession’ refers to a single palm in the entire collection, and the term ‘entry’ refers to an accession selected for the inclusion in the core collection. The cluster-based stratified sampling strategy was used to establish the core collection (van Hintum et al., Reference van Hintum, Brown, Spillane and Hodgkin2000; Zewdie et al., Reference Zewdie, Tong and Bosland2004), which included two steps. The first step involved the determination of clusters based on the following two methods: cluster analysis and sorting method. The cluster analysis was carried out based on Nei and Li's (Reference Nei and Li1979) coefficient of genetic similarity and the unweighted pair group method with arithmetic mean (UPGMA) clustering method using the NTSYSpc software (Rohlf, Reference Rohlf2000). The sorting method consisted of a multiple correspondence analysis (MCA) using the coefficient of genetic similarity based on the proportion of shared alleles (PS), according to the formula proposed by Bowcock et al. (Reference Bowcock, Ruiz-Linares, Tomfohrde, Minch, Kidd and Cavalli-Sforza1994):
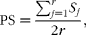 $$\begin{eqnarray} PS = \frac { \sum _{ j = 1}^{ r } S _{ j }}{2 r }, \end{eqnarray}$$
$$\begin{eqnarray} PS = \frac { \sum _{ j = 1}^{ r } S _{ j }}{2 r }, \end{eqnarray}$$
where S j is the number of shared alleles by two individuals at locus j (j= 1 to r).
The second step was the selection of individuals to determine the number of entries to be included in each cluster, which constituted the core collection. Therefore, three methods were employed for the selection of entries. Two of these methods have been proposed previously by Brown (Reference Brown1989), who stated that the number of accessions within clusters is based on the proportion (P) or the logarithmic (L) ratio of the size of each cluster in the whole collection. Finally, for the third selection method of entries, which contains all genotypic features in the core collection, a heuristic algorithm was used as proposed by Kim et al. (Reference Kim, Chung, Cho, Ma, Chandrabalan, Gwag, Kim, Cho and Park2007). This analysis was carried out using the (PowerCore software, http://www.genebank.go.kr/eng/PowerCore/powercore.jsp/). Indicators of genetic diversity in the whole and core collections were calculated using the (GENALEX software, http://biology.anu.edu.au/GenALEx/; Peakall and Smouse, Reference Peakall and Smouse2006). Genetic diversity was quantified by the number of alleles (A) and expected heterozygosity (H e) retained in the core collection with respect to the whole collection.
Size of the core collection
Brown (Reference Brown1989), Spagnoletti (Reference Spagnoletti and Qualset1993), Li et al. (Reference Li, Zhang, Zeng, Yang, Shen, Sun and Wang2002), Zewdie et al. (Reference Zewdie, Tong and Bosland2004) and Yan et al. (Reference Yan, Rutger, Bryant, Bockelman, Fjellstrom, Chen, Tai and McClung2007) suggested that 10% of the entire collection could be an acceptable sample size for the core collection, and that this fraction could contain 70% of the total diversity. In this study, according to the sampling strategies P and L, the core collection contained 78 oil palm accessions (E. guineensis Jacq.) that constituted a sample size of 10%.
Results
Allelic diversity
The SSR molecular markers used in this study were polymorphic among the accessions evaluated, producing a total of 271 alleles (Table 1). The number of alleles identified in the analysed SSR loci ranged from 2 (sEg00126) to 18 (mEgCIR3362). Table 1 shows the SSR loci ordered according to the expected heterozygosity (H e), where the first locus (mEgCIR3362) was most informative and most likely to detect the differences between two individuals, and the last marker (sEg00126) was least informative and least likely to detect the differences. The mEgCIR3362 locus exhibited the highest values for allelic richness (R a = 13.69), observed heterozygosity (H o = 0.82) and expected heterozygosity (H e = 0.90). Of the identified alleles, 30% corresponded to the private alleles. Here, the term ‘private alleles’ refers exclusively to the alleles that were present in only one of the three countries. The 455 accessions of oil palm from Angola contained 36 private alleles with frequencies ranging from 0.001 to 0.188; the 311 accessions of oil palm from Cameroon contained 26 private alleles with frequencies ranging from 0.002 to 0.080; and the 22 accessions of oil palm from Indonesia contained 20 private alleles, of which 6 were identified using the mEgCIR3543 locus. The frequencies of those 20 private alleles ranged from 0.023 to 1.000, indicating that despite the limited number of oil palm accessions analysed, the samples from Indonesia exhibited the highest number of private alleles.
Table 1 Allelic diversity in microsatellite loci evaluated in oil palm accessions (Elaeis guineensis)

n, number of palms; R a, allelic richness; H o, observed heterozygosity; H e, expected heterozygosity; PI, probability of identity.
Genetic differentiation and diversity
According to Nei's (Reference Nei1987) coefficients of genetic diversity and differentiation, a low coefficient of genetic differentiation (G ST= 0.022) was observed between the oil palm accessions from Angola and those from Cameroon, which exhibited a total genetic diversity (H T) value of 0.658 (Table 2). The distribution pattern of the accessions obtained from the MCA did not reflect a separation in terms of the geographical origins from which the palms were collected (Fig. 1). The three dimensions of the MCA explained 100% of the variation in the accessions analysed, in which each dimension explained 33.3% of the variation. This lead to the occurrence of potential introgression between the accessions from Angola and those from Cameroon, and to low genetic differentiation between them. The inclusion of the 22 accessions from Indonesia in the analysis resulted in a high coefficient of genetic differentiation (G ST= 0.178–0.251), indicating that the accessions from Indonesia were genetically different from those acquired from Africa (Angola and Cameroon). This analysis was carried out using the same number of accessions for all geographical origins (22 accessions per origin selected randomly), exhibiting a G ST value of 0.249, which also indicated a high coefficient of genetic differentiation between these accessions. The oil palm accessions from Cameroon and Indonesia exhibited the highest genetic diversity (H T= 0.804). The distribution of genotypes within each group of accessions was measured by the inbreeding coefficient (G IS). The values obtained were low (close to zero), positive and significant (0.054 < G IS>0.170) (Table 2), showing that the allelic frequencies of the genotypes evaluated were within the expected proportions that were consistent with Hardy–Weinberg equilibrium.
Table 2 Nei's coefficients of genetic diversity and differentiation between oil palm accessions
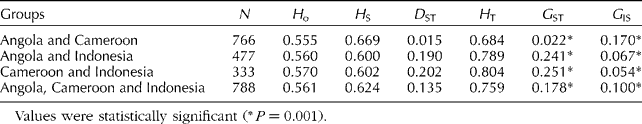
N, number of entries; H o, average of observed genetic diversity; H S, average of genetic diversity within the groups; H T, total genetic diversity; D ST, average of genetic diversity among the subgroups; G ST, coefficient of genetic differentiation; G IS, inbreeding coefficient.
Values were statistically significant (* P= 0.001).

Fig. 1 Three-dimensional (Dim) multiple correspondence analysis of 766 oil palm accessions from Angola and Cameroon.
Establishment of the core collection
Determination of clusters
The dendrogram generated by NTSYSpc using the UPGMA clustering method showed the formation of many clusters with few accessions (see online supplementary Fig. S1). Therefore, we could not identify the distinguishable groups clearly, as the cluster analysis did not determine the optimal number of clusters. Nevertheless, the MCA determined the optimal number of clusters by evaluating the similarities among the individuals based on the χ2 distances. Thus, the results of the MCA revealed the establishment of 11 groups for 766 accessions (Fig. 2). Cluster I represents 77% of the accessions from Cameroon. Clusters II and III collectively represent 84% of the accessions from Angola. Overall, 80% of the accessions were distributed in clusters I, II and III. Although clusters X and XI represent only one accession each, we decided to include these entries in the core collection. The oil palm accessions from Indonesia were not included in the cluster-based stratified sampling because they may mask the few differences between the accessions from Angola and those from Cameroon; instead, these accessions were considered as an additional cluster (cluster XII; see Table 2).

Fig. 2 Three-dimensional (Dim) multiple correspondence analysis showing the classification of 766 oil palm accessions from Angola and Cameroon into 11 clusters.
Allocation of the number of entries in each cluster
A new structure for the whole collection was established, which was divided into 12 clusters. Three sampling methods were used for the allocation of the number of entries. The number of alleles (A) and the expected heterozygosity (H e) were determined for each group and for the total number of the entries selected. However, only the total number of entries was used to quantify the genetic diversity that could be included in the core collection (Table 3). According to the P and L strategies, approximately 235 alleles could be included in a sample that represented 10% of the entire collection. The L strategy was more effective for the retention of alleles and a high genetic diversity value in clusters with fewer accessions. Two subsets were established using the (PowerCore software, http://www.genebank.go.kr/eng/PowerCore/powercore.jsp/). In the first subset, the entries were selected from each group. As a result, 289 entries were selected, which would be the retention of 271 alleles in a sample representing 37% of the entire collection. In the second subset, the entries were selected from the 788 accessions contained within the whole collection, regardless of the clusters established previously. As a result, 91 entries were selected, representing the 12 groups established previously by the MCA and containing 271 retained alleles with a total H e value of 0.72 in a sample representing 11% of the entire collection.
Table 3 Number of entries selected from each cluster using several sampling strategies

N, number of entries; A, number of alleles; H e, expected heterozygosity (Nei, Reference Nei1987).
a Entries were selected from each group.
b Entries were selected from 788 accessions.
Finally, fully random sampling from a different set of SSRs was used (Table 4), with the aim of comparing the methods that have been used previously. For set 1, the total number of SSRs was used. For set 2, the following SSRs were used: mEgCIR0067; mEgCIR0219; mEgCIR0230; mEgCIR1772; sEg00125; sEg00126; sEg00127; mEgCIR0173; mEgCIR3282; mEgCIR3292; mEgCIR3543; mEgCIR3546; mEgCIR3785; mEgCIR3886. For set 3, the following SSRs were used: mEgCIR0008; mEgCIR0009; mEgCIR0221; mEgCIR0254; mEgCIR0437; sEg00066; sEg00067; sEg00090; sEg00140; mEgCIR0802; mEgCIR1730; mEgCIR1753; mEgCIR3292; mEgCIR3362. Consequently, the number of alleles (A) and the expected heterozygosity (H e) were less in the three random sets than in the PowerCore, P and L strategies.
Table 4 Number of entries selected fully randomly from a different set of simple sequence repeats (SSRs)
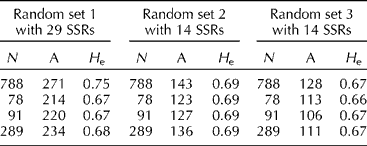
N, number of entries; A, number of alleles; H e, expected heterozygosity (Nei, Reference Nei1987).
Discussion
The accessions of oil palm (E. guineensis) evaluated in this study exhibited significant genetic diversity for this species. Therefore, these accessions are considered as a useful genetic resource for increasing the genetic base of oil palm breeding populations. The introduction of new germplasm sources is crucial to the development of new varieties with competitive advantages for the plant breeding sector. A total of 271 alleles were obtained from the 29 SSRs evaluated and private alleles were identified for each geographical origin, which was reflected in the high genetic diversity values obtained for the 788 oil palm accessions evaluated. Similar genetic diversity values for oil palms (E. guineensis) have been reported previously by Billotte et al. (Reference Billotte, Risterucci, Barcelos, Noyer, Amblard and Baurens2001), Montoya et al., (Reference Montoya, Arias, Rey and Rocha2005), Singh et al. (Reference Singh, Mohd, Ting, Rosli, Tan, Leslie, Ithnin and Cheah2008), Arias et al. (Reference Arias, Montoya and Romero2013a, Reference Arias, González, Prada, Restrepo and Romerob). Although microsatellites have been demonstrated to be versatile molecular markers for genetic diversity analysis in oil palms, it is important to consider that they may impose some limitations on data analysis. Microsatellites generally reflect the repeat size ranging from 2 to 5 bp of DNA. In practice, the occurrence of other alleles that correspond to changes outside of the repeat unit is likely, leading to other variations in repeat size, including single-base differences in some microsatellites. The detection of such differences would be difficult with the gel system used in this study, for which the resolution is around 2 bp, which could complicate the interpretation of microsatellite allele frequencies and thus make the estimates of relatedness faulty.
The parameters of genetic differentiation and the MCA determined in this study revealed a low coefficient of genetic differentiation between the populations of palms collected from Angola and Cameroon. This finding may be attributed to the geographical distribution of oil palms in Africa and their continuous expansion since the early days of the slave trade from the coasts of Cape Verde to Angola, which maintained active seed dispersal, prevented the structuring of populations and promoted genetic diversity of the species (Corley and Tinker, Reference Corley and Tinker2003). The results of the present study are consistent with those reported previously by Kularatne et al. (Reference Kularatne, Shah and Rajanaidu2001); Billote et al. (Reference Billotte, Risterucci, Barcelos, Noyer, Amblard and Baurens2001); Barcelos et al. (Reference Barcelos, Amblard, Berthaud and Seguin2002); Hayati et al. (Reference Hayati, Wickneswari, Maizura and Rajanaidu2004), Montoya et al., (Reference Montoya, Arias, Rey and Rocha2005); Maizura et al. (Reference Maizura, Rajanaidu, Zakri and Cheah2006), Arias et al. (Reference Arias, Montoya and Romero2013a, Reference Arias, González, Prada, Restrepo and Romerob). These previous studies using different types of molecular markers have reported that, oil palm accessions from different African countries did not exhibit a specific and distinct genetic structure, which was attributed predominantly to the dispersion of the material without any geographical barriers over the African continent. Conversely, Nei's (Reference Nei1987) coefficients of genetic differentiation indicated that Deli dura accessions from Indonesia are genetically distinct from oil palm accessions from Africa. Furthermore, these accessions exhibited the greatest number of private alleles compared with those of oil palm from Africa. Although many factors or biological phenomena are known to alter allelic frequencies, the genetic differences found among all the accessions could be attributed to natural processes and some historical events. Historical evidence shows that the oil palm is of African origin, and that its distribution along the equatorial tropics has been the result of the process of domestication by humans. In 1848, four Deli dura palms from the Bogor Botanical Gardens were introduced to the Botanical Gardens of Amsterdam and Mauritius. However, the exact origin of these palms remains unknown (Corley and Tinker, Reference Corley and Tinker2003). When oil palms were introduced to Southeast Asia, the conditions in Northern Sumatra were found to be more favourable for increasing the production of bunches, the average weight of bunches and the potential of oil production (Gerritsma and Wessel, Reference Gerritsma and Wessel1997). The results of the present study are consistent with those reported previously by Kularatne et al. (Reference Kularatne, Shah and Rajanaidu2001), who evaluated oil palm accessions collected from different geographical origins using the amplified fragment length polymorphism (AFLP) markers. The study by Kularatne et al. (Reference Kularatne, Shah and Rajanaidu2001) found that Deli dura accessions are significantly different from oil palm populations in Africa, indicating that oil palm accessions from Indonesia and Africa can be treated as separate management units, as they have accumulated significant genetic differences between them.
The initial purpose of this study was to develop a core collection using molecular data to maximize the representativeness of genetic diversity. This will certainly allow the efficient use of genetic resources and, at the same time, a reduction in the costs of evaluation and characterization. The core collection can be modified eventually to accommodate new knowledge and new diversity. Cluster-based stratified sampling is one of the most effective methods for the establishment of a core collection, as the variation within clusters is minimized and the variation between clusters is maximized (Diwan et al., Reference Diwan, McIntosh and Bauchan1995; Cochran, Reference Cochran1977). One of the most commonly used clustering methods is the UPGMA method. However, in this study, clearly differentiable clusters could not be identified. Therefore, clustering criteria were established based on a sorting method similar to that shown in the MCA. Results obtained from clustering analysis can be unstable when many clustered individuals are intermediate in terms of their similarity and/or differences compared with the others. In this case, these individuals are not assigned to a specific cluster, which leads either to the formation of several clusters consisting of only a few individuals or to individuals that are not assigned to any cluster at all. This problem can be overcome by using sorting methods because they do not group individuals according to a hierarchical structure. However, these methods may identify the relationships between individuals based on the presence or absence of a common molecular marker. After the identification of the relationship, the array of the individuals according to their similarities or differences can be determined easily. The MCA represents the similarities between individuals based on the χ2 distances. This is the most appropriate approach for the analysis of molecular data, as it is sufficiently sensitive for detecting the patterns of similarity based on rare alleles that are shared by some genotypes (Laurentin, Reference Laurentin2009). The MCA was the most suitable approach for defining the clusters that contain the total genetic diversity and for more effectively establishing an oil palm core collection that is representative of the genetic diversity of the whole collection. The results of this study show that the P strategy was more effective in retaining alleles within clusters with the largest number of accessions, whereas the L strategy was more effective in retaining alleles within clusters with fewer accessions. The two methods used for assigning the number of entries retained the maximum number of alleles and the observed genetic diversity within a sample representing 10% of the entire collection. This finding is consistent with that reported previously by Brown (Reference Brown1989), who established that these two methods optimize the entry allocation process within a core collection. However, the combination of stratified sampling based on a sorting method and a heuristic algorithm developed by Kim et al. (Reference Kim, Chung, Cho, Ma, Chandrabalan, Gwag, Kim, Cho and Park2007) was found to be the most effective method than the P and L strategies for the development of an oil palm core collection set while maintaining allele numbers and genetic diversity. Furthermore, based on the accuracy of the classification of individuals, PowerCore proved to be a powerful tool for streamlining the generation process of the core collection set of oil palms. Therefore, based on the allelic diversity evaluated using the 29 SSR loci in 788 oil palm accessions, two core collections were identified. The first core collection consisted of 289 entries (37% of the entire collection), while the second core collection is a mini core collection consisting of 91 entries (11% of the entire collection). The results of this study are consistent with those reported previously by Chung et al. (Reference Chung, Kim, Chung, Lee, Lee, Dixit, Kang, Zhao, McNally, Hamilton, Gwag and Park2009), who found that the heuristic method used for establishing a core collection was more efficient than the P and random core collection strategies. Zhao et al. (Reference Zhao, Cho, Ma, Chung, Gwag and Park2010) found similar results for the development of a rice core collection. The use of the heuristic method was significantly more favourable than the random sampling method used for capturing the maximum number of alleles with minimum redundancy. Moe et al. (Reference Moe, Gwag and Park2012) previously demonstrated the efficiency of PowerCore in the development of a core collection using AFLP markers in mung bean plants.
The oil palm is an ideal example of a crop whose yield still responds to improved environments and whose genetic potential has not been fully exploited. Its adaptation to new environments represents a process that will continue to produce diversification according to new demands for domestication. Thus, accessions collected from Indonesia and Africa are a valuable genetic resource that may represent a long-term benefit for the development of resistance to various biotic and abiotic factors. We proposed the development of an oil palm core collection using molecular tools as a measure to increase its economical use and to develop strategies for the conservation of genetic resources. This core collection would be of great interest to oil palm researchers because new strategies for breeding programmes can be developed based on these advances to increase production and introduce new varieties that exhibit excellent nutritional qualities and are resistant to pests, diseases, drought and salinity.
Supplementary material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/S1479262114001026
Acknowledgements
The authors thank the Angolan National Coffee Institute and the Institute for Agricultural Research and Development (IARD) for their collaboration and permission to collect the oil palm material. They thank Dr Leonardo Rey for collecting the oil palm material in Africa. They also thank Myriam Cristina Duque and Juan Bosco, researchers at the International Center for Tropical Agriculture (CIAT), for their assistance in the data analysis. This study was funded by the Oil Palm Development Fund (FFP), managed by Fedepalma.








