Introduction
Foxtail millet (Setaria italica (L.) P. Beauv.) (2n = 18), is one among the world's most important ancient domesticated crops. Its domestication in China dates back to 8700 years ago (Lu et al., Reference Lu, Zhang, Liu, Wu, Li, Zhou, Ye, Zhang, Zhang, Yang, Shen, Xu and Li2009). It is widely cultivated in Asia, Europe, North America, Australia and North Africa as grain food or forage (Austin, Reference Austin2006). Taxonomically, foxtail millet comprises two subspecies, S. italica subsp. italica (foxtail millet) and subsp. viridis (green millet). Green millet is considered as the wild ancestor of foxtail millet (Kihara and Kishimoto, Reference Kihara and Kishimoto1942; Li et al., Reference Li, Li and Pao1945). Based on the comparative morphology, Prasada Rao et al. (Reference Prasada Rao, de Wet, Brink and Mengesha1987) have recognized three races of foxtail millet; Moharia, Maxima and Indica.
Foxtail millet has gradually become a minor crop in the last 80 years due to the rapid development of maize and other crops (Austin, Reference Austin2006). Poor seedling establishment, need for hand weeding, lack of breeding effort for improvement (Ahanchede et al., Reference Ahanchede, Hamon and Darmency2004) and lack of proper utilization of genetic variability for the development of improved cultivar are other reasons for its neglect. Therefore, phenotypic and molecular characterization of existing plant genetic resources is essential for proper utilization in crop improvement.
The simple sequence repeat (SSR) markers are now the markers of choice in most areas of plant genetics and breeding due to many desirable attributes including hypervariability, multiallelic nature, co-dominant inheritance, reproducibility, relative abundance, extensive genome coverage, chromosome-specific location, amenability to automation and high-throughput genotyping (Kalia et al., Reference Kalia, Rai, Kalia, Singh and Dhawan2011). SSR markers were used to study genetic diversity in several crops (Hokanson et al., Reference Hokanson, Szewc-McFadden, Lamboy and McFerson1998; Hao et al., Reference Hao, Zhang, Wang, Dong, Shang and Jia2006; Kottapalli et al., Reference Kottapalli, Burow, Burow, Burke and Puppala2007; Stępień et al., Reference Stępień, Mohler, Bocianowski and Koczyk2007; Ali et al., Reference Ali, Rajewski, Baenziger, Gill, Kskridge and Dweikat2008; Upadhyaya et al., Reference Upadhyaya, Dwivedi, Baum, Varshney, Udupa, Gowda, Hoisinton and Singh2008b; Agrama et al., Reference Agrama, Yan, Lee, Fjellstrom, Chen, Jia and McClung2009; Blair et al., Reference Blair, Díaz, Buendía and Duque2009; Koppolu et al., Reference Koppolu, Upadhyaya, Dwivedi, Hoisington and Vershney2010; Zhang et al., Reference Zhang, Zhang, Wang, Sun, Qi, Li, Wei, Han, Qiu, Tang and Li2011). However, molecular or biochemical characterization of an entire germplasm collection is unrealistic (Gepts, Reference Gepts1995) because of the large size of germplasm collection conserved in genebanks for many crops. This could be more easily fulfilled by using subsets of the whole collection (Liu et al., Reference Liu, Sun, Salomon and Bothmer2002). Core collections (Frankel, Reference Frankel1984) are subsets consisting about 10% of the entire collection and represent the diversity of the entire collection of the species. Upadhyaya et al. (Reference Upadhyaya, Pundir, Gowda, Reddy and Singh2008a) developed the core collection of foxtail millet (155 accessions) from an entire collection of 1474 accessions using the data on taxonomic and qualitative traits. Extensive molecular characterization of this foxtail millet core collection could provide information about diversity parameters, allelic richness, genetic relationship among the races, population structure and marker–trait associations. It also provides useful information for selecting the most diverse accessions that could be used as parents for making crosses to study the segregating generations as well as the development of mapping populations. Therefore, the objectives of the study were (1) to estimate genetic diversity and allelic richness in the foxtail millet core collection using SSR markers, (2) to study the relationship among the three races in foxtail millet and (3) to identify the most diverse accessions for use in breeding programmes.
Materials and methods
Plant materials and genomic DNA extraction
The genetic material used in this study consisted of 155 accessions of ICRISAT foxtail millet core collection (Upadhyaya et al., Reference Upadhyaya, Pundir, Gowda, Reddy and Singh2008a) which includes all three races and ten sub-races from 23 countries. It consisted of 102 accessions from the race Indica (65.8%), 24 accessions from the race Maxima (15.5%) and 29 accessions from the race Moharia (18.7%). The foxtail millet core collection was planted in the 3rd week of October 2009 in a glass house at ICRISAT, Patancheru, India and DNA was extracted from the single seedling of each of the 155 accessions by a high-throughput 96-well mini-DNA extraction method (Mace et al., Reference Mace, Buhariwalla and Crouch2003).
SSR genotyping
At the start of the investigation, there were no SSR markers publicly available for foxtail millet except few EST-SSRs (Jia et al., Reference Jia, Shi, Song, Wang, Wang and Li2007). Initially, four foxtail millet EST-SSRs (Jia et al., Reference Jia, Shi, Song, Wang, Wang and Li2007), 31 pearl millet (Thudi et al., Reference Thudi, Senthilvel, Bottley, Hash, Reddy, Feltus, Paterson, Hoisington and Varshney2010) and 24 finger millet (Dida et al., Reference Dida, Srinivasachary, Ramakrishnan, Bennetzen, Gale and Devos2007) genomic SSR markers were used. Later, a set of 80 SSR markers located across nine chromosomes of foxtail millet were selected based on the foxtail millet linkage map reported by Jia et al. (Reference Jia, Zhang, Liu, Zhang, Shi, Song, Wang and Li2009). The forward primers of all these 139 SSR markers were synthesized by adding the M13 forward primer sequence (5′-CACGACGTTGTAAAACGAC-3′) at the 5′end of each primer. All these 139 markers were tested for amplification using a set of eight phenotypically most diverse accessions (ISe 31, ISe 746, ISe 748, ISe 827, ISe 995, ISe 1037, ISe 1129 and ISe 1227) in the core collection for optimizing PCR conditions and the concentration of the reaction mix, and to check polymorphism. Finally, three foxtail millet EST-SSRs, 12 finger millet SSRs, four pearl millet SSRs and all 80 foxtail millet SSRs were identified as polymorphic on the eight diverse accessions. Thus, a total of 99 SSR markers were used for genotyping the entire foxtail millet core collection.
Genomic DNA of all the genotypes were diluted to 5 ng/μl and used as a template for the amplification of the SSR markers. PCRs were performed in 5 μl volume consisting of 1 μl of 5 ng DNA template, 0.5 μl of 2 mM dNTPs, 0.2 μl of 25 mM MgCl2, 0.5 μl of the primer containing 1:5:5 a ratio of 2 pmol/μl M13-tailed forward primer, 2 pmol/μl reverse primer and 2 pmol/μl of either 6-Fam, Vic, Ned or Pet (Applied Biosystems), 0.5 μl of 10 × PCR buffer and 0.15 U of Taq DNA polymerase (SibEnzymes Ltd, Russia). PCR amplifications were performed on an ABI thermal cycler (GeneAmp, PCR system 9700; Applied Biosystems) using a common touchdown PCR amplification profile for the series of markers. The touchdown PCR amplification profile was 94°C for 3 min of initial denaturation cycle, followed by first ten cycles of 94°C for 15 s, 61°C for 30 s and 72°C for 30 s, with a 1°C decrease in temperature per cycle, then 40 cycles of 94°C for 15 s with a constant annealing temperature (54°C) and 72°C for 30 s, followed by a final extension at 72°C for 20 min. The PCR products were tested for amplification on 1.2% agarose.
Capillary electrophoresis
A set of 25 PCR multiplex were constructed based on the allele size range estimates and the type of forward primer label of the markers. Each set consisted of four SSR markers with different labels and allele size. For post-PCR multiplexing, 1 μl PCR product of each of 6-Fam, Vic, Ned and Pet-labelled products were pooled (according to the above-mentioned criteria) and mixed with 7 μl of Hi-Di formamide (Applied Biosystems, USA), 0.2 μl of the LIZ-500 size standard (Applied Biosystems, USA) and 2.8 μl of distilled water. The pooled PCR amplicons were denatured for 5 min at 95°C and cooled immediately on ice and size-separated by capillary electrophoresis using an ABI Prism 3730xl DNA analyser (Applied Biosystems, Inc.). Raw data produced from the ABI 3730xl DNA analyser were analysed using Genemapper® software version 4.0 (Applied Biosystems, USA) and fragment size was scored in base pairs based on the relative migration of the internal size standard, LIZ 500.
Molecular data analysis
The fragment sizes for all markers were used to analysis basic statistics using PowerMarker version 3.25 (Liu and Muse, Reference Liu and Muse2005), including the polymorphic information content (PIC), allelic richness as determined by the total number of the detected alleles and the number of alleles per locus, gene diversity and the occurrence of unique, rare, common and most frequent alleles and heterozygosity (%). Unique alleles are those that are present in one accession or in one group of accessions but absent in other accessions or group of accessions. Rare alleles are those whose frequency is ≤ 1% in the investigated materials. Common alleles are those occurring between 1 and 20% in the investigated materials while those occurring >20% were classified as most frequent alleles (Upadhyaya et al., Reference Upadhyaya, Dwivedi, Baum, Varshney, Udupa, Gowda, Hoisinton and Singh2008b). The correlations among the number of repeat unit, the number of alleles per locus, gene diversity, heterozygosity and PIC were estimated.
A neighbour-joining tree was constructed based on the simple matching dissimilarity matrix of 84 polymorphic markers genotyped across the foxtail millet core collection as implemented in DARwin 5.0.156 program (Perrier and Jacquemoud-Collet, Reference Perrier and Jacquemoud-Collet2006). Most diverse pairs of accessions were identified based on the dissimilarity matrix. The principal coordinate analysis (PCoA) was performed based on Nei's (Reference Nei1973) distance matrix using GENALEX 6.41 (Peakall and Smouse, Reference Peakall and Smouse2006). Pairwise F st and genetic distance among the three races were calculated using GENALEX 6.41. Analysis of molecular variance (AMOVA) was performed to partition the molecular variance within and between the races and populations identified by the cluster analysis based on 999 permutations using the software GENALEX 6.41.
Results
Initially, 135 SSRs and four EST-SSRs were used to check polymorphism across eight phenotypically most diverse accessions in the foxtail millet core collection. Of these, 99 markers including 80 SSR foxtail millet SSR, three unmapped foxtail millet EST-SSR, 12 finger millet SSR and four pearl millet SSR markers were used for genotyping the entire core collection accessions. Finally, 84 markers (72 foxtail millet SSRs, three foxtail millet EST-SSRs, five finger millet SSRs and four pearl millet SSRs) produced clear, scorable and polymorphic marker profiles and were therefore used for further analysis.
Molecular diversity in the foxtail millet core collection
A set of 84 markers (81 SSRs and 3 EST-SSRs) detected a total of 1356 alleles in the 155 accessions of foxtail millet core collection (Table 1). Of the 1356 alleles, 368 were rare alleles (27.1%), 906 common alleles (66.8%) and 82 the most frequent alleles (6.1%). P5 and p92 had no rare alleles, whereas for all other markers, rare alleles ranged from 1 (p61, b202, b196, b226, p34, p269, p56, p20, p21, b115, m2, ICMM02C05, P2 and UGEP102) to 12 (ICMM02C24 and UGEP81) (see Supplementary Table S1, available online only at http://journals.cambridge.org). A total of 61 unique alleles that were present only in one accession and absent in the other accessions were detected. The number of alleles per locus ranged from 4 (b196 and p56) to 35 (b171) with an average of 16.14 alleles per locus. The PIC value is a reflection of allele diversity and the informativeness of each marker. The PIC values ranged from 0.06 (b196) to 0.95 (b260) with an average of 0.70. Of the 84 markers, 75 showed a PIC value >0.30 and were highly informative and polymorphic. Gene diversity is defined as the probability that two randomly chosen alleles from the population are different. It varied from 0.06 (b196) to 0.95 (b260), with an average of 0.72. Of the 84 SSRs, 71 showed high gene diversity (>0.50) and only 13 SSR markers scored gene diversity ≤ 0.50 (Table 1; see Supplementary Table S1, available online only at http://journals.cambridge.org).
Table 1 Summary statistics based on the three races and the entire foxtail millet core collection using the 84 simple sequence repeat locia

PIC, polymorphic information content.
a Values in parentheses represent the range.
The mean heterozygosity was very less in the entire core collection (0.06). Of the 84 markers used, ten SSR markers detected no heterozygosity and 11 SSR markers showed >0.10 (Table 1; see Supplementary Table S1, available online only at http://journals.cambridge.org). Of these 11 SSR markers, three each for foxtail millet EST-SSRs (P5, P13 and P2), finger millet SSRs (UGEP102, UGEP3 and UGEP56) and pearl millet SSRs (ICMMO2D15B, ICMMO2C24 and ICMM02D07) showed heterozygosity >0.10. Of the 72 foxtail millet SSR markers used, only two SSRs [p16 (0.16) and p2 (0.22)] showed heterozygosity >0.10.
Molecular diversity among the races in the foxtail millet core collection
Biologically, the accessions of foxtail millet core collection belonged to three races namely Indica (102 accessions), Maxima (24 accessions) and Moharia (29 accessions). Of the 1356 alleles detected in the entire core collection, 997 (73.5%) alleles were detected in the race Indica, 784 (57.8%) in Maxima and 844 (62.2%) in Moharia (Table 1). Of the 1356 alleles, 43% of alleles were present in all three races called shared alleles. Of the 997 alleles present in accessions from the race Indica, 100 (10.1%) were rare alleles, 803 (80.5%) common alleles and 94 (9.4%) the most frequent alleles. Of the 784 alleles in Maxima, 688 (87.8%) were common alleles and 96 (12.2%) the most frequent alleles. In the case of Moharia, 760 (90.1%) common alleles and 84 (9.9%) most frequent alleles were detected out of the 844 alleles. Rare alleles were not detected in accessions from the races Maxima and Moharia but were found only in the race Indica. This is due to the less sample size of Maxima and Moharia when compared with Indica in the core collection. A total of 44 unique alleles in Indica, 77 unique alleles in Maxima and 47 alleles in Moharia were detected.
A similar range of PIC values was found in all the three races, which ranged from 0.02 to 0.93 in Indica, 0.08 to 0.91 in Maxima and 0.12 to 0.94 in Moharia with an average of 0.63 in Indica and 0.72 in Maxima and Moharia. For Indica, gene diversity averaged 0.65, ranging from 0.02 to 0.93, whereas the accessions from Maxima varied from 0.08 to 0.92 with an average of 0.74. In Moharia, gene diversity ranged from 0.13 to 0.94 with an average 0.74. Maxima and Moharia had the maximum mean gene diversity and PIC than Indica. The average PIC values and gene diversity in the races showed the highest range, which indicated the diversity of the three biological races present in the core collection and highly informative SSR markers used.
Genetic relationship among the races
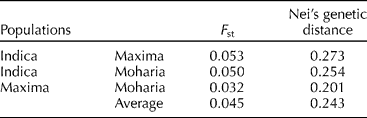
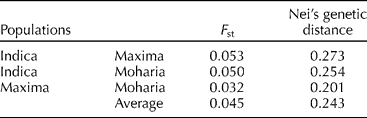
Pairwise comparison based on the F st values of could be interpreted as the standardized population distance between two populations, i.e., the proportion of the total genetic diversity that separates the population. F st is typically greater than or equal to zero. If all the subpopulations are in Hardy–Weinberg equilibrium with the same allele frequency, then F st = 0. In this study, the overall pairwise F st among the three races were 0.045 (Table 2). Maxima showed the smallest F st with Moharia (0.032), whereas Indica showed the maximum F st with Maxima (0.053) and Moharia (0.050). The genetic distance data are in agreement with the F st estimates. Indica showed the highest genetic distance with Maxima (0.273) and Moharia (0.254), whereas the genetic distance between Maxima and Moharia was the smallest (0.201).
Table 2 Pairwise estimates of F st and Nei's genetic distance based on the 84 simple sequence repeat loci among the three races
The neighbour-joining tree was constructed based on simple matching dissimilarity matrix of the 155 accessions in the foxtail millet core collection using the DARwin 5.0.156 program, which highlighted broadly four clusters named as cluster I (C-I) to cluster IV (C-IV) (Fig. 1). Colour coding was given based on three biological races, namely Indica (red), Maxima (green) and Moharia (blue). C-I contained 87 accessions, of which 80 accessions were from Indica. C-II consisted of 35 accessions. Of these 35 accessions, the majority (22) of the accessions were from Indica with few accessions from Maxima (seven) and Moharia (six). C-III was mainly represented by Moharia (17 accessions out of 19 accessions present in this cluster). C-IV was represented by Maxima predominantly with 11 accessions. C-I and C-II were represented by accessions from Indica, and C-III and C-IV were dominated by accessions from Moharia and Maxima, respectively.

Fig. 1 Neighbour-joining tree based on the simple matching dissimilarity matrix of the 84 SSR markers genotyped across the 155 accessions of the foxtail millet core collection (colour coding based on the three races). A colour version of this figure can be found online at journals.cambridge.org/pgr
The neighbour-joining clustering pattern was further assessed using PCoA. The first three PCos explained 66.2% of the variation, of which PCo1 explained 34.7% of the variation and PCo2 explained 16.6% of the SSR variation among the 155 accessions in the foxtail millet core collection (Fig. 2). Plotting the first two PCos shows clear separation of accessions of Indica, whereas Maxima and Moharia were not clearly separated.

Fig. 2 Principal coordinate (PCo) analysis of the foxtail millet core collection accessions using the 84 SSR markers based on Nei's (Reference Nei1973) distance estimates.
Analysis of molecular variance
AMOVAs were conducted to determine the structure of genetic diversity and the variation explained within and between the three races and four clusters (Table 3). Only a relatively small portion (7%) of the molecular variation was explained by the races while 93% was among the individuals within the races. A similar pattern was observed when the AMOVA estimated based on the four clusters (10% among the clusters and 90% within the clusters) indicated the consistent relationship of the clusters with the races, a high degree of variability within each group and a low level of genetic variation among the races.
Table 3 Analysis of molecular variance based on the three races and four clusters identified by the neighbour-joining tree using the 84 simple sequence repeat markers

Correlations
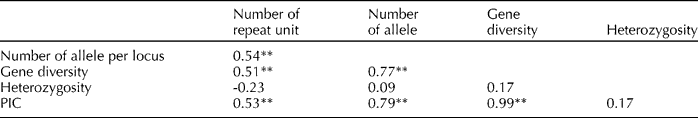
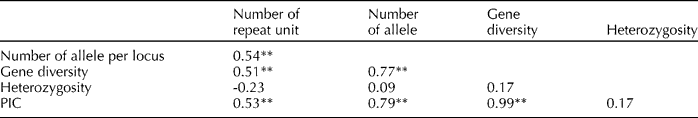
The correlation coefficients among the number of repeat unit, the number of alleles per locus, gene diversity, heterozygosity and PIC for 84 SSR markers were estimated to study the relationship between the above parameters (Table 4). The number of repeat unit was highly significant and positively correlated with the number of alleles per locus (0.54), gene diversity (0.51) and PIC (0.53). The number of alleles per locus was highly significant and positively correlated with gene diversity (0.77) and PIC (0.79), and gene diversity was highly significant and positively correlated with PIC (0.99).
Table 4 Correlations among the number of repeat unit, the number of alleles per locus, gene diversity, heterozygosity and polymorphic information content
PIC, polymorphic information content.
** indicate significance at the 0.05 and 0.01 levels, respectively.
Genetically diverse accessions
The dissimilarity matrix based on SSR polymorphism was used to determine the level of relatedness among the accessions present in the core collection. Pairwise estimates of dissimilarity values ranged from 0.098 to 0.956. The maximum dissimilarity (0.956) was observed between ISe 1320 (Moharia) and ISe 1162 (Indica). Based on the dissimilarity values, 250 pairs of the most diverse accessions were identified in the foxtail millet core collection (see Supplementary Table S2, available online only at http://journals.cambridge.org) (for the dissimilarity matrix of the 155 accessions based on the 84 SSR markers, see Supplementary Table S3, available online only at http://journals.cambridge.org). Moreover, the pairwise estimates of dissimilarity values were >0.50 except for 123 pairs of the accessions out of 11,935 pairwise estimates indicating a high level of diversity prevailing in the ICRISAT foxtail millet core collection.
Discussion
Success of a crop improvement programme depends on the knowledge and availability of genetic variability present in that crop for efficient conservation, management and effective utilization of plant genetic resources (Mondini et al., Reference Mondini, Noorani and Pagnotta2009). Foxtail millet is as an important crop and attracts international research attention due to its high drought tolerance, photosynthesis efficiency (Liu et al., Reference Liu, Bai, Zhang, Znu, Xia, Cheng and Shi2011), nutritional values and health benefits. With a small diploid genome (~400 Mb; Bennetzen et al., Reference Bennetzen, Schmutz, Wang, Percifield, Hawkins, Pontaroli, Estep, Feng, Vaughn, Grimwood, Jenkins, Barry, Lindquist, Hellsten, Deshpande, Wang, Wu, Mitros, Triplett, Yang, Chu-Yu, Mauro-Herrera, Wang, Li, Sharma, Sharma, Ronald, Panaud, Kellogg, Brutnell, Doust, Tuskan, Rokhsar and Devos2012), C4 panicoid crop and short duration, foxtail millet has become a model system for studying biofuel crops and comparative genomics among the grasses (Wang et al., Reference Wang, Chen, Zhi, Yang, Li, Wang, Li, Zhao, Chen and Diao2010; Bennetzen et al., Reference Bennetzen, Schmutz, Wang, Percifield, Hawkins, Pontaroli, Estep, Feng, Vaughn, Grimwood, Jenkins, Barry, Lindquist, Hellsten, Deshpande, Wang, Wu, Mitros, Triplett, Yang, Chu-Yu, Mauro-Herrera, Wang, Li, Sharma, Sharma, Ronald, Panaud, Kellogg, Brutnell, Doust, Tuskan, Rokhsar and Devos2012). Due to the reduced size of foxtail millet core collection from the entire collection of 1474 accessions, molecular characterization of foxtail millet core collection helps breeders to use genetic resources for cultivar development more effectively.
Our results showed that there was a large molecular diversity in the foxtail millet core collection. The SSR markers used in this study were highly polymorphic and informative, and detected a total of 1356 alleles with an average of 16.14 alleles per locus. The number of alleles per locus ranged from 4 to 35, which was higher than that reported in barley (4–32 alleles; Matus and Hayes, Reference Matus and Hayes2002) and rice (3–32 alleles; Borba et al., Reference Borba, Brondani, Breseghello, Coelho, Mendonça, Rangel and Brondani2010), and less than that reported in chickpea (14–67 alleles; Upadhyaya et al., Reference Upadhyaya, Dwivedi, Baum, Varshney, Udupa, Gowda, Hoisinton and Singh2008b) and maize (2–38 alleles; Wang et al., Reference Wang, Yu, Zhao, Shi, Song, Wang and Li2008). The average of 16.14 alleles per locus was detected in the foxtail millet core collection, which was more than that reported by previous studies in foxtail millet (6.16; Jia et al., Reference Jia, Zhang, Liu, Zhang, Shi, Song, Wang and Li2009; 14.04, Liu et al., Reference Liu, Bai, Zhang, Znu, Xia, Cheng and Shi2011; 2.4, Lin et al., Reference Lin, Liao, Chiang, Kuoh and Chang2012) and other crops, e.g. 7.6 (Wang et al., Reference Wang, Zhu, Barkley, Chen, Erpelding, Murray, Tuinstra, Tesso, Pederson and Yu2009) and 4.79 (Shehzad et al., Reference Shehzad, Iwata and Okuno2009) in sorghum, 8.23 in maize (Yang et al., Reference Yang, Yan, Shah, Warburton, Li, Li, Chai, Fu, Zhou, Xu, Bai, Meng, Zheng and Li2010) and 8.2 (Agrama et al., Reference Agrama, Eizenga and Yan2007), 15.8 (Agrama and Eizenga, Reference Agrama and Eizenga2008) and 12.4 (Borba et al., Reference Borba, Brondani, Breseghello, Coelho, Mendonça, Rangel and Brondani2010) in rice. Jia et al. (Reference Jia, Zhang, Liu, Zhang, Shi, Song, Wang and Li2009) used only 40 accessions of foxtail millet from limited geographical origins (most from China), which might be the reason for the less number of alleles per locus in their study. In the foxtail millet core collection, 368 were rare alleles (27.14%), 906 common alleles (66.81%) and 82 the most frequent alleles (6.05%). In general, markers detecting a greater number of alleles per locus detected more rare alleles. Rare alleles from cultivated and wild accessions could be used to select specific accessions for allele mining (Upadhyaya et al., Reference Upadhyaya, Yadav, Dronavalli, Gowda and Singh2010). A total of 61 unique alleles were detected in the core collection, which were specific to a particular accession. Unique alleles are important because they may be diagnostic of a particular type of genotype (Senior et al., Reference Senior, Murphy, Goodman and Stuber1998) and useful for cultivar identification. The presence of many rare and unique alleles could be due to the higher rate of mutation at SSR loci (Henderson and Peters, Reference Henderson and Peters1992; Senior et al., Reference Senior, Murphy, Goodman and Stuber1998) and a highly diverse nature of the accessions present in the ICRISAT foxtail millet core collection. PIC in this study was higher (0.70) than that reported in sweet sorghum (0.54; Wang et al., Reference Wang, Zhu, Barkley, Chen, Erpelding, Murray, Tuinstra, Tesso, Pederson and Yu2009) and rice (0.42; Jin et al., Reference Jin, Lu, Xiao, Sun, Corke and Bao2010; 0.603, Pervaiz et al., Reference Pervaiz, Rabbani, Pearce and Malik2009), but lower than that reported in chickpea (0.854; Upadhyaya et al., Reference Upadhyaya, Dwivedi, Baum, Varshney, Udupa, Gowda, Hoisinton and Singh2008b). The maximum allelic richness and the maximum number of rare and unique alleles can be explained by several factors such as diversity range of the germplasm, number of accessions used, number of SSR loci and SSR repeat type. A larger number of SSR loci and the use of dinucleotide repeat SSRs rather than trinucleotide or higher may lead to a higher number of alleles and higher genetic diversity (Yang et al., Reference Yang, Yan, Shah, Warburton, Li, Li, Chai, Fu, Zhou, Xu, Bai, Meng, Zheng and Li2010). The SSR markers used in this study are dinucleotides, which might be one of the reasons for higher allelic diversity. Moreover, the higher number of alleles may also be attributed to the material used in this study; core collection represents the diversity of the entire collection of foxtail millet conserved at ICRISAT, Patancheru, India, which is an indication of the greater diversity present in this core collection and its potential as a reservoir of novel alleles for crop improvement. Although foxtail millet is a self-pollinating crop and DNA was extracted from single representative plant of each accession, some SSR loci showed relatively more heterozygosity (>10%), especially three foxtail millet EST-SSRs, two genomic SSRs and SSRs from other crop/related species (pearl millet and finger millet). This could be due to outcrossing averaging about 4% (Li et al., Reference Li, Meng and Liu1935) in foxtail millet or heterozygous individuals, or by residual heterozygosity in germplasm (Blair et al., Reference Blair, Díaz, Buendía and Duque2009) for particular SSR loci.
This study is the first to report the estimates of genetic diversity and the relationship within and among the three races recognized by Prasada Rao et al. (Reference Prasada Rao, de Wet, Brink and Mengesha1987). Of the 1356 alleles detected in the entire core collection, 73.5% alleles were detected in the race Indica, 57.8% in Maxima and 62.2% in Moharia. Even though the number of accessions in Maxima and Moharia was less than that in Indica, these two races had more than 55% of total alleles present in the entire core collection, indicating the maximum diversity of these two races as well. Accessions of Moharia often resemble the members of wild ssp. viridis in phenotype, except that they have lost the ability of natural seed dispersal (Prasada Rao et al., Reference Prasada Rao, de Wet, Brink and Mengesha1987), hence it might have harboured more diversity. The higher number of alleles in Indica might also be due to the more number of accessions represented in the core collection (65.8%) when compared with Maxima (15.5%) and Moharia (18.7%). The maximum number of unique alleles was detected in Maxima and Moharia when compared with Indica. This could be due to the less number of sample size when compared with Indica. Here, the molecular-based biological (races) diversity differed with respect to allelic richness, frequency of rare, common and most frequent alleles, which can be explained by the difference in sample size included in each race and genetic diversity harboured in it. AMOVA based on the three races and four clusters explained high within-population diversity. This is in agreement with previous studies in different crops (Abdurakhmonov et al., Reference Abdurakhmonov, Kohel, Yu, Pepper, Abdullaev, Kushanov, Salakhutdinov, Buriev, Saha, Scheffler, Jenkins and Abdukarimov2008; Jun et al., Reference Jun, Van, Kim, Lee and Walker2008; Peleg et al., Reference Peleg, Fahima, Abbo, Krugman and Saranga2008a, Reference Peleg, Saranga, Krugman, Abbo, Nevo and Fahimab). On the basis of neighbour-joining clustering, F st, genetic distance and PCoA to visualize the genetic relationship among the races, we concluded that the molecular diversity of foxtail millet was fairly correlated well with racial classification and the race Indica showed a greater genetic distance from the races Maxima and Moharia. On the basis of the multi-environment evaluation of the foxtail millet core collection (data not shown), we noted that the accessions belonging to Indica were taller, flowered late and had a greater flag leaf blade length, flag leaf sheath length, peduncle length, inflorescence length, single plant yield and plot yield than those from Maxima and Moharia. Prasada Rao et al. (Reference Prasada Rao, de Wet, Brink and Mengesha1987) reported that a high number of basal tillers (5–52, average 8.6) is a characteristic feature of the race Moharia. The accessions of Maxima were intermediate for days to flowering, plant height, flag leaf blade length, flag leaf sheath length, peduncle length, inflorescence length, inflorescence width, single plant yield and plot yield. The 155 accessions of the foxtail millet core collection were from 23 countries, with the majority being from India, and a few accessions were from other countries. We did not obtain a clear-cut grouping of the accessions based on either countries or regions of origin.
In summary, the results of this study provide evidence for the presence of a high level of genetic diversity in the ICRISAT foxtail millet core collection as well as within and between the three races. This foxtail millet core collection also possesses a good amount of diversity for important agronomic traits (Upadhyaya et al., Reference Upadhyaya, Pundir, Gowda, Reddy and Singh2008a). Here we have identified 250 pairs of most diverse accessions showing a high level of marker diversity (see Supplementary Table S2, available online only at http://journals.cambridge.org). These pairs of accession could be used by other researchers and breeders as parents in their breeding programmes for making crosses to identify and (or) to study superior segregants, and for the development of mapping populations. These SSR markers were useful in differentiating closely related germplasm sources and races in foxtail millet. Hence, these SSR markers could be used to supplement morphological and agronomic data used for the protection of plant variety and/or germplasm identification. The ICRISAT foxtail millet core collection accessions are conserved at ICRISAT genebank and available to the researchers following the Standard Material Transfer Agreement.
Acknowledgements
The authors gratefully acknowledge the financial support of the BMZ/GTZ project on ‘Sustainable conservation and utilization of genetic resources of two underutilized crops – finger millet and foxtail millet – to enhance productivity, nutrition and income in Africa and Asia’ funded by the Federal Ministry for Economic Cooperation and Development (BMZ), Germany.