INTRODUCTION
Trypanosoma (Megatrypanum) theileri parasitic in cattle is the type species of the subgenus T. (Megatrypanum) (Hoare, 1972), a heterogeneous group of large trypanosomes infecting almost all mammalian orders (Hoare, 1972; Wells, 1976). Molecular phylogenies have indicated that the subgenus T. (Megatrypanum) is polyphyletic (Stevens et al. 1999, 2001). Hoare (1972) suggested that morphologically similar trypanosomes infecting domestic and wild ruminants should all be classified as T. theileri, and proposed the abolition of several synonyms of T. theileri created for trypanosomes infecting different Bovidae species and cattle breeds. Isolates from Cervidae species have either been classified as T. theileri or as separate species based on morphology (Kingston and Morton, 1975; Dirie et al. 1990a,b; Bose et al. 1993).
T. theileri trypanosomes share mammalian host restriction, worldwide distribution, contaminative transmission by tabanids and lack of pathogenicity. All attempts to infect mammals other than cattle with T. theileri have failed (Hoare, 1972; Wells, 1976). A T. theileri-like parasite of cervids was not infective to cattle (Kingston and Morton, 1975) and bovid T. theileri were not infective to deer (Bose et al. 1987). We showed that T. theileri trypanosomes from cattle and water buffalo living together clustered according to host species by RAPD analysis, suggesting some degree of host specificity of these trypanosomes (Rodrigues et al. 2003).
T. theileri is distributed worldwide from the tropics to near the Arctic Circle, with higher prevalence in tropical and neotropical areas. Trypanosomes morphologically indistinguishable from one another and from T. theileri have been described infecting Bovidae, Antilocapridae, Tragulidae and Cervidae with cattle accounting for the largest number of reports (Hoare, 1972; Wells, 1976; Schlafer, 1979; Rodrigues et al. 2003).
T. theileri is considered non-pathogenic, causing a chronic and cryptic infection usually only detectable in blood cultures. Many cases indicate its potential pathogenicity in concurrent infections with Babesia, Theileria, Anaplasma or piroplasms, or associated with stress, poor nutrition and gestation (Wells, 1976; Hussain et al. 1985; Braun et al. 2002). All trypanosomes that are pathogenic to artiodactyls are of African origin and positioned in the ‘T. brucei clade’, cyclically transmitted by tsetse flies and mechanically by other biting flies. T. theileri and ‘T. brucei clade’ trypanosomes share artiodactyl host species, have overlapping distributions and bovids carrying mixed trypanosome infection are commonly found in the field. All the ‘T. brucei clade’ trypanosomes show broad host-specificity, infecting a much wider range of vertebrates than their ‘natural’ host range, i.e. the hosts they evolved in, which were probably wild artiodactyls, e.g. suids and antelopes (Logan-Henfrey et al. 1992; Gibson et al. 2001; Stevens et al. 2001). In Latin America, cattle, buffalo and cervids have been reported infected by T. theileri and related species (Hoare, 1972; Wells, 1976; Rodrigues et al. 2003). ‘T. brucei group’ parasites have been imported into Latin America where they are always mechanically transmitted; T. vivax is widespread in cattle (Ventura et al. 2001); T. evansi infects horses, dogs and wild mammals (Ventura et al. 2002) and cattle (Herrera et al. 2004).
Tabanidae (Diptera) flies, which are thought to be the most important vectors of T. theileri occur worldwide and are also mechanical vectors for salivarian trypanosomes (Hoare, 1972; Krinsky, 1976; Foil, 1989). Development of T. theileri in tabanids is typical of stercorarian trypanosomes; i.e., restricted to the gut with transmission by penetration of metacyclic trypomastigotes in the faeces through the bite wound of the vector, mucosa, or by ingestion of faeces or of the vector itself by the host (Wells, 1976; Bose et al. 1987; Bose and Heister, 1993). The presence of metacyclic trypomastigotes in the salivary glands suggests that ticks can transmit T. theileri and T. cervi by bite, a route peculiar to salivarian trypanosomes (Krinsky and Burgdorfer, 1976; Morzaria et al. 1986).
Phylogenetic relationships among T. theileri trypanosomes have never been addressed in the previous studies (Stevens et al. 1999, 2001; Hamilton et al. 2005) by comparing several isolates from different host species and geographical origin. In this work, we analysed SSU and ITS ribosomal sequences of these trypanosomes aiming (a) to infer phylogenetic relationships among isolates from water buffalo, never included before in phylogenetic studies, with isolates from cattle and fallow deer, (b) to assess the genetic polymorphism among Brazilian isolates from cattle and water buffalo from distant geographical regions and (c) to analyse relationships of these non-pathogenic with pathogenic trypanosomes parasitic in artiodactyls.
MATERIALS AND METHODS
Trypanosoma spp. used in this study, isolation and growth of T. theileri isolates
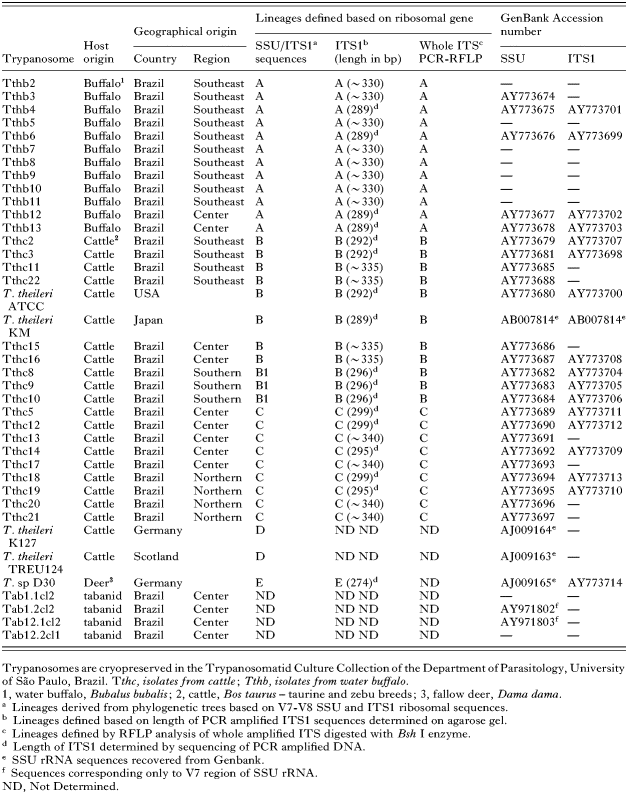
In this study we compared 34 T. theileri isolates, 18 from cattle (Bos taurus) and 12 from water buffaloes (Bubalus bubalis), most isolates (30) came from Brazilian regions: (a) North, State of Rondônia (RO), Amazonia; (b) Central, Mato Grosso do Sul State (MS), Pantanal; (c) Southeast, São Paulo State (SP); (d) Southern, Paraná (PR) and (e) Rio Grande do Sul (RS) States (Table 1; Fig. 2). The largest geographical distance (~3·600 km) is between RO and RS (Fig. 2B). Trypanosomes from water buffaloes were from Southeast (SP) and Central (Pantanal, MS) regions. Most cattle isolates were recovered from zebu-cattle (Nelore breed, Indian origin), except some isolates from SP and PR States, which are from taurine-cattle (Holstein breed, European origin), and the isolates from RO State, which are all from cross-breed (zebu and taurine) cattle. Trypanosomes were isolated and cultured as before (Rodrigues et al. 2003). Trypanosoma sp D30 was isolated from the fallow deer Dama dama in Germany (Bose et al. 1993).

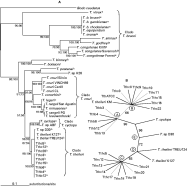
Fig. 1 (A) Phylogenetic tree of 39 trypanosome species from artiodactyls and several other mammalian orders based on Maximum Likelihood analysis of SSU rRNA (V7-V8) sequences: a, pathogenic species parasites of artiodactyls of clade Trypanosoma brucei; b, non-pathogenic species parasites of artiodactyls of clade T. theileri; c, T. (Megatrypanum) spp. from non-artiodactyl hosts (−lnL 5642.1548). The numbers at nodes correspond to percentage of bootstrap values (Maximum Likelihood: Maximum Parsimony) derived from 100 replicates. (B) Maximum Likelihood analysis of SSU rRNA (V7-V8) sequences from 27 isolates of the clade T. theileri. T. cyclops was used as outgroup for clade T. theileri (−lnL 1385.6146). The numbers correspond to percentage bootstrap support values from 100 replicates. A-E corresponds to the 5 lineages of isolates disclosed by analysis of clade T. theileri.

Fig. 2. (A) Phylogeny based on Maximum Likelihood analysis using ITS1 rDNA sequences from isolates representative of all lineages (excepting D) disclosed using SSU rRNA sequences) (Fig. 1) of Trypanosoma theileri from cattle (B and C), and T. theileri-related trypanosomes from water buffalo (A) and fallow deer (E) previously defined using SSU rRNA sequences (Fig. 1). T. rangeli was used as outgroup. Symbols on each isolate correspond to geographical origin of T. theileri. (B) Geographical origin of Brazilian isolates, Southeast ([bull ]), South ([utrif ]), Central ([squf ]) and North ([lozf ]) regions. North American (○), European ([loz ]) and Japanese (□) isolates. The numbers correspond to percentage of ML bootstrap support values derived from 100 replicates.

PCR amplification, sequencing of SSU and ITS ribosomal genes, and restriction analysis
DNA of cultured trypanosomes was obtained by classical phenol-chloroform extraction. A ~900 bp DNA fragment containing V7-V8 regions of SSU rRNA gene was PCR amplified and products from 3 independent reactions were pooled and automatically sequenced as before (Maia da Silva et al. 2004). DNA fragments corresponding to whole ITS rDNA (ITS1, 5·8S and ITS2 sequences) or to only ITS1 rDNA sequences were obtained by PCR, cloned and 2–3 clones from each isolate were sequenced as before (Maia da Silva et al. 2004). For restriction analysis, whole ITS amplified DNA were digested with Bsh I enzyme and analysed on 2·0% agarose gel stained with ethidium bromide. DNA of T. vivax, T. b. brucei and T. evansi were obtained as before (Ventura et al. 2001, 2002). Nucleotide sequence data reported in this paper are available in the GenBank database under the Accession numbers listed in the Table 1.
DNA preparation and PCR amplification of DNA from T. theileri trypanosomes infecting tabanids
Crude preparations of DNA templates were obtained from individual digestive tract of dried preserved specimens of Tabanus sp using the method described by Aljanabi and Martinez (1997). Tabanids feeding on cattle and horses were caught using small nets in 2 regions from where T. theileri isolates were also obtained: MS (24 specimens) and SP (12) States. All DNA preparations were submitted to T. theileri specific PCR-Tth625 assay (Rodrigues et al. 2003) and samples positive in this assay were used as template for amplification of the V7 region of the SSU rRNA (~500 bp) using the primers 609F and 1156R (Maia da Silva et al. 2004), with annealing temperature of 56 °C and adding 5 μg of BSA to reaction mixtures. Probably due to poor quality of DNA templates, only small DNA fragments (V7SSU rRNA) could be amplified. PCR products were cloned and 3–5 clones from each sample were sequenced as before (Maia da Silva et al. 2004).
Alignment of SSU and ITS1 ribosomal sequences and phylogenetic inferences
Partial SSU rRNA sequences (730 bp of V7-V8 regions) from T. theileri trypanosomes determined in this study were aligned with sequences of cattle isolates from Japan and Europe, and with the sequence from the cervid trypanosome (D30) from GenBank (Table 1). Sequences from other trypanosomes from artiodactyls were also retrieved from GenBank (Accession number): T. b. brucei (AL359782); T. b. gambiense (AJ009141); T. b. rhodesiense (AJ009142); T. vivax (U22316); T. congolense Kilifi (AJ009144); T. congolense Savannah (AJ009146); T. congo-lense Forest (AJ009145); T. simiae (AJ009162); T. godfreyi (AJ009155); T. equiperdum (AJ009153); T. evansi (D89527). We also included in the alignment sequences from trypanosomes of several other mammalian orders (GenBank): T. pestanai (AJ009159); T. sp H26 (AJ009169); T. binneyi (AJ132351); T. cruzi VINCH89 (AJ009149); T. cruzi Sylvio X10 (AJ009147); T. cruzi CLBR (AF245383); T. cruzi CanIII (AJ009148); T. rangeli PG (AJ012416); T. rangeli San Augustin (AJ012417); T. minasense (AJ012413); T. leeuwenhoeki (AJ012412); T. legeri (AY491769); T. conorhini (AJ012411); T. cyclops (AJ250743); T. sp ABF (AJ620564). The sequence of T. boissoni (U39580) from fish was also included in the alignment and the sequence of Bodo caudatus was used as outgroup for Trypanosoma.
Alignments were made using as guide the general alignment of the rRNA database (http://rrna.uia.ac.be/) with subsequent manual adjustment. Maximum parsimony (MP) and maximum-likelihood (ML) analysis were based on 1162 well-aligned characters and carried out using PAUP 4.0b10. The ML model and parameters were estimated using the hierarchical likelihood test implemented in the program Modeltest, version 3.06 (Posada and Crandall, 1998). The best-fit evolutionary model for the likelihood analysis determined by Modeltest was Tamura and Nei with gamma distribution shape. MP and ML analysis and bootstrapping with 100 replicates for both analyses were done as described (Hamilton et al. 2004, 2005). Previous analysis demonstrated that phylogenetic trees inferred using V7-V8 sequences of SSU rRNA disclosed the same branching pattern and all major clades of trypanosomes (Maia da Silva et al. 2004) compared to trees generated using larger SSU rRNA sequences (Hamilton et al. 2004). Alignments used in this study are available from the authors upon request.
A second alignment was done to analyse the clade T. theileri using 17 V7-V8 sequences of SSU rRNA from Brazilian isolates of T. theileri from cattle (17 isolates) and water buffaloes (5 isolates), plus 5 isolates from cattle from USA, Japan, Germany and Scotland (Table 1). Sequences corresponding to V7-SSU rRNA (minimum 500 bp) obtained from crude preparations of trypanosomes infecting tabanids, all from MS State, were aligned with sequences from trypanosomes of clades T. theileri and T. cyclops, the last including 2 trypanosomes from wallaby, T. sp ABF (GenBank Accession number AJ620564) and T. sp 10 (AJ620571), and 1 isolate from leech, TL.AV.43cl1 (AJ620571). Restricting the alignment to closely related trypanosomes allowed characters in more variable regions to be aligned with higher confidence and included in the ML analysis (K80 evolutionary model) of these very closely related taxa.
Alignment of ITS sequences, similarity matrices and phylogeny (ML) were done as before (Maia da Silva et al. 2004). The evolutionary model determined by Modeltest was F81 with gamma distribution shape. Alignment did not reveal significant polymorphism among ITS1 sequences of clones from the same isolate.
RESULTS
Phylogeny of T. theileri and related trypanosomes based on SSU rRNA sequences
Phylogenetic relationships among trypanosomes parasitic in Artiodactyla and species infecting different mammalian orders were inferred by comparison of SSU rRNA sequences (V7-V8 regions). We analysed 27 T. theileri isolates and related trypanosomes and 11 isolates of 7 species of salivarian trypanosomes parasites of artiodactyls: T. vivax; T. congolense; T. b. brucei; T. evansi; T. equiperdum; T. simiae; T. godfreyi. The large genetic distances among the several trypanosome species used to infer the phylogeny of Trypanosoma, contrasted with the homogeneity of T. theileri trypanosomes prevented visualization of the branching pattern within the T. theileri clade. For this reason, after analysis using all isolates (data not shown) we selected 12 representatives of all major groups to represent the T. theileri clade in the phylogeny of Trypanosoma (Fig. 1A). Species from all major clades of mammalian trypanosomes, parasites of almost all other mammalian orders, were included in this analysis. Members of the T. theileri clade were tightly clustered into a strongly supported (100% bootstrap) monophyletic assemblage. Similar strongly supported topologies were obtained for phylogenetic trees inferred using both ML (Fig. 1A) and MP (data not shown) methods and the bootstrap values varied only slightly (Fig. 1A). The clade T. theileri was separated from all other trypanosomes of the subgenus T. (Megatrypanum) infecting other mammalian orders (T. conorhini, T. legeri, T. minasense, T. pestanai, and T. binneyi). The T. theileri clade was closest to T. cyclops (~89% similarity) and T. cruzi (~87% similarity). The ‘T. brucei clade’, comprising all salivarian trypanosomes, was very distinct from the ‘T. theileri clade’.
Grouping of T. theileri trypanosomes according to polymorphism on the SSU ribosomal sequences
To clarify the branching pattern within the T. theileri clade and the phylogenetic relationships among the closely related trypanosomes of this clade (Fig. 1A), 17 sequences from Brazilian isolates of T. theileri from cattle and 5 from buffaloes were compared with sequences from cattle isolates from USA, Japan, Germany and Scotland (Table 1). T. cyclops was used as outgroup. Despite the high similarity (99·9–97·8%), the SSU rRNA (V7-V8 regions) segregated isolates of T. theileri clade into 2 major subclades (Fig. 1B). One subclade was formed by 2 clusters comprising isolates from water buffaloes (lineage A) and from cattle (lineage B). The second subclade was subdivided in 3 branches (lineages C, D and E). Lineages C and D contain only cattle isolates and lineage E 1 isolate from deer (Fig. 1B). Thus, the 5 isolates from water buffaloes (lineage A) were tightly clustered together (100% similarity) whereas Brazilian isolates from cattle were divided in 2 clusters (lineages B and C) separated from European cattle isolates (D). The sequence divergence among the 3 groups of cattle isolates ranged from 0·5 to 1·2%, with higher heterogeneity within lineage C. The trypanosome from deer (D30), the only sample available from Cervidae, was on a separate branch (lineage E) closest to isolates from European cattle (Fig. 1).
Congruence of lineages defined by analysis of polymorphism of ITS and SSU ribosomal sequences
Length and sequence polymorphisms in the ITS rDNA sequences were analysed to evaluate the consistency of grouping within the clade T. theileri. The aligned whole ITS of the Brazilian cattle (Tthc3) and buffalo (Tthb6) isolates with sequences from cattle isolates from USA and Japan showed significant sequence divergence on ITS1 (6·1%) and ITS2 (2·6%), whereas sequences of 5·8S were identical.
To compare phylogenies of T. theileri and related species using sequences with different evolutionary rates, highly conserved SSU were compared with more variable ITS1 sequences from isolates representative of clusters previously defined by SSUrRNA analysis (Fig. 1B). Isolates from lineage D (Europe) were not available to be included in this analysis. The topology of the ITS1 dendrogram (Fig. 2A) was the same as that generated by SSU analyses (Fig. 1B), with total congruency of topologies using both MP (data not shown) and ML methods, showing the same two major monophyletic assemblages of isolates, one formed for A and B, and the other for C, D and E branches.
In agreement with the SSU rRNA data, lineages A and B showed the smallest distance based on ITS1 sequences whereas the largest distances separated lineages A and C (Fig. 2A). In contrast to the homogeneity (100% similarity) among buffalo isolates (lineage A), a significant polymorphism was showed by cattle isolates within both lineages B and C. Lineage B showed 2·7% of divergence intra-lineage and was divided into 2 sublineages: B1, consisted of isolates from the Southern region sharing 100% of similarity, and B2, comprising isolates from the Southeast region also sharing 100% similarity, plus the isolates from USA and Japan, the most divergent within this lineage. Lineage C was the most heterogeneous, with intra-lineage divergence ranging from 1·2% to 5·5% (average of 3·3%).
Considering the average sequence divergence among isolates of the same host species, cattle isolates were separated from water buffalo and cervid isolates by 26% and 35%, respectively, whereas isolates from buffalo were separated from the cervid isolate by 38% of sequence divergence. Besides significant similarity between isolates from cattle and buffalo (lineages A and B) living together, the largest genetic distances separated Brazilian cattle isolates from North (C) and South (B1) Brazil (Fig. 1B) suggested higher divergence between more geographically distant populations or independent introductions. The existence in Pantanal (Central Brazil) of the lineages detected in Southeast (SP) and Northern (RO) regions (Table 1, Figs 1 and 2A) is consistent with relative geographical distances (Fig. 2B) and the known regular transport of cattle among these regions.
Detection and sequence analysis of trypanosomes of T. theileri clade infecting tabanids
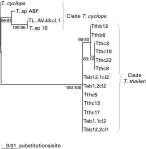
Tabanid specimens positive for T. theileri and related trypanosomes were screened using T. theileri-specific PCR-Tth625 that generated a 500 bp DNA band specific for these trypanosomes (Rodrigues et al. 2003). Of 36 tabanid specimens, 22 were positive for T. theileri. DNA fragments corresponding to the V7 region of SSU rRNA from 6 specimens positive for T. theileri trypanosomes selected by PCR-Tth625 were cloned and at least 3 clones were sequenced for each sample. Alignment of 13 sequences of very good quality, obtained from 6 tabanid specimens from MS State, and sharing greatest (minimum 98%) similarity to T. theileri according to BLAST searches, disclosed small polymorphism with the 4 different sequences obtained represented by the following clones: Tab1.1cl2, Tab1.2cl2, Tab12.1cl2, Tab12.2cl1. All selected DNA sequences from tabanid specimens were tightly clustered in the clade T. theileri (100% of bootstrap) (Fig. 3).

Fig. 3. Relationships among Trypanosoma theileri trypanosomes from vertebrate host, cattle (Tthc) and water buffalo (Tthb), and invertebrate (Tabanus spp) (Tab) hosts based on alignment of V7SSU rRNA sequences using ML analysis. Bootstrap values (Maximum Likelihood: Maximum Parsimony) were derived from 100 replicates.
Length and restriction fragment length polymorphism of ITS rDNA as a tool for diagnosis and genotyping of trypanosomes of clade T. theileri
To standardize a method for easy identification and lineage typing of T. theileri trypanosomes, without DNA sequencing and only requiring a small amount of DNA, we evaluated (a) PCR amplification of whole ITS or ITS1 sequences and (b) PCR-RFLP of ITS DNA. The length of PCR-amplified whole ITS or ITS1 of trypanosomes from cattle, buffalo and fallow deer was similar in 2% agarose gels, and is consequently suitable for differentiation of members of the T. theileri clade from other trypanosomes parasitic in ruminants (Fig. 4). The difference in size of PCR product was insufficient to reliably identify T. theileri groups by size alone; however, it was possible to discriminate T. theileri from the other species tested by size (Fig. 4; Table 1). Three banding patterns were identified in the PCR-RFLP gels, each one corresponding to one lineage previously defined for T. theileri of Brazilian cattle (B and C) and buffalo (A) isolates (Fig. 4). The restriction pattern of Trypanosoma sp (D30) from deer differed from those of cattle and buffalo isolates (data not shown).

Fig. 4. Agarose gels (2%) stained with ethidium bromide (Etbr) of amplified ITS rDNA sequences from isolates of Trypanosoma theileri and allied species and salivarian species of trypanosome parasitic in ruminants. PCR-wITS, length polymorphism of PCR amplified whole ITS DNA sequence (ITS1+5·8S+ITS2) and PCR-ITS1, exclusive amplification of ITS1. Negative controls were done using a mixture of DNA from cattle and water buffalo. PCR-RFLP, analysis of PCR amplified whole ITS DNA from trypanosomes of clade T. theileri, selected to illustrate distinct lineages within clade T. theileri, digested with Bsh I enzyme used for lineage genotyping.
DISCUSSION
Phylogenetic relationships among trypanosomes parasitic in artiodactyls were inferred for non-pathogenic T. theileri from cattle, related trypanosomes from water buffalo and fallow deer, and pathogenic trypanosomes parasitic in artiodactyls (African trypanosomes). T. theileri and allied species were tightly clustered into a well-supported T. theileri clade by analysis of SSU and ITS DNA sequences, and were distinct from all T. (Megatrypanum) of other mammalian orders, confirming the previously reported polyphyly of this subgenus (Stevens et al. 1999, 2001). Since T. theileri is the type species of T. (Megatrypanum) (Hoare, 1972) the name T. (Megatrypanum) should only apply to members of the T. theileri clade and not to other species that have previously been included in this subgenus.
Divergence on SSU and ITS rDNA sequences separated trypanosomes of artiodactyls of clade T. theileri into 5 lineages. Isolates from the three host species examined in this study never clustered together, even when they were collected from the same locations, suggesting a high degree of host specificity. Lineage A was related to water buffalo, lineages B, C and D to cattle and lineage E to fallow deer. However, the significance of host-parasite associations must be interpreted with caution, particularly when the number of isolates examined from different host species is small and when experimental tests of host-specificity have not been carried out. Although cattle and water buffalo generally live in dry and swampy areas, respectively, segregation of trypanosomes from these hosts living on the same farms and sharing the same pastures indicated lack of host switching and is consistent with host restriction of these trypanosomes. We have never found animals with mixed lineage infections (Rodrigues et al. 2003). Although only one isolate from deer was available for this study, it was recognized as a separated lineage (E) based on data from this and from previous studies consistent with host specificity (Kingston and Morton, 1975; Krinsky, 1976; Dirie et al. 1990a,b; Bose et al. 1993). The molecular data showing distinct groups of parasites in different species is consistent with the lack of cross-infectivity in experimental studies in field conditions, all biochemical studies and different trypanosomes isolated from distinct artiodactyl species, while those from the same hosts were indistinguishable (Dirie et al. 1990a,b; Bose et al. 1993; Rodrigues et al. 2003).
Only trypanosomes of Bovidae and Cervidae, artiodactyls of the Pecora suborder (former Ruminantia), were positioned in the clade T. theileri. There are some old reports of these trypanosomes in chevrotain (ruminant of the suborder Tragulina) and camels (Tylopoda, primitive ruminants) (cited in Wells, 1976). However recent surveys using morphological and molecular approaches detected salivarian trypanosomes in camels and suids (Gibson et al. 2001), but never T. theileri. Broader analysis of isolates from other artiodactyls is required to determine the range of vertebrate host species of the T. theileri clade.
Small sequence divergence indicated that host-associated lineages are recently diverged and could be correlated to host phylogeny (Hassanin and Douzery, 1999). Isolates from cattle and buffaloes (Bovidae) are more related to each other than to Cervidae isolate. Lineage B of cattle trypanosomes and lineage A of buffalo isolates share the same geographical regions. Lineage B showed to be more related to lineage A than to other lineages of cattle parasites from more distant regions. Zebu cattle and water buffaloes are both from India and recently introduced in Brazil. Only analysis of trypanosomes from Indian water buffaloes could clarify if differences arose before these animals arrived in Brazil and the relationships between isolates from these hosts. Asian (Bubalus bubalis) and African (Syncerus caffer) buffaloes are infected by T. theileri-like, and although these trypanosomes from buffaloes were never compared, both differed from cattle isolates (Dirie et al. 1990a; Rodrigues et al. 2003). The large genetic distance separating artiodactyl trypanosomes suggests an ancient divergence of T. theileri and the T. brucei clade. Artiodactyls originated in the northern hemisphere in the early Eocene (54,8–33,7 mybp) and by the end of the Oligocene had radiated into a number of families (Hassanin and Douzery, 1999). All data suggested that T. theileri trypanosomes are recently diverged, compatible with a recent divergence of their host species. T. theileri probably evolved with ruminant artiodactyls and dispersed with the artiodactyl radiation in the Miocene and, more recently, by humans transporting livestock around the world.
Our results using a T. theileri-specific PCR assay (Rodrigues et al. 2003) indicated a high prevalence of T. theileri trypanosomes infecting tabanids, thus confirming for the first time using a molecular method the role of these flies as important invertebrate hosts and, probably, as the most important vectors of these trypanosomes. Sequences of V7SSU rRNA of trypanosomes infecting tabanids were tightly clustered together within the clade T. theileri. Moreover, these sequences clustered together with those of cattle isolates from the same geographical region (MS State). Tabanids may have been in existence since the Eocene/Oligocene (Martins-Neto, 2003). The developmental cycle of T. theileri in tabanids and transmission ability of these flies had been well demonstrated through experimental infections (Wells, 1976; Bose et al. 1987; Bose and Heister, 1993). The data strongly suggest that T. theileri of bovids are transmitted by tabanid species, which are catholic feeders, whereas host-specific hippoboscid flies are important vectors for cervid trypanosomes (Wells, 1976; Foil, 1989; Bose et al, 1993). We are currently analysing a larger number of tabanids from distinct regions in order to understand vector-parasite relationships within the T. theileri clade.
The T. theileri clade is clearly a complex taxon comprising lineages associated with host species and geographical origin, and is constituted by different populations that appear to have evolved with non-random association of independent molecular markers (clonal structure). However, more biological and molecular data from isolates from different artiodactyls, from widespread geographical origins, will be required for a fuller investigation of host-parasite relationships and to determine segregation factors of T. theileri lineages.
We are grateful to Vania L. Nunes, Arlete Dell`Porto, Ivete Conchon and Maria Cristina C. Carollo for help with bovid blood samples. We also thank Gentilda F. Takeda and several undergraduate students for inestimable help in the fieldwork. Work in Rondônia (Monte Negro) was done at the laboratory of the ICBV-USP. DNA of T. congolense and Trypanosoma sp D30 were kindly supplied by Professor A. Tait (Glasgow, UK). We thank Wendy C. Gibson for critical comments on the manuscript. This work was supported by the Brazilian agencies FAPESP and CNPq. Adriana C. Rodrigues is a CNPq student fellow.