



Introduction
In vitro embryo production (IVP) is a reproductive technology used to increase the number of offspring from genetically elite animals and to study early embryo development in different species of mammals (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016). Although researchers have had considerable success in increasing IVP efficiency, the quality of the produced embryos remains lower than those produced in vivo (Lonergan and Fair, Reference Lonergan and Fair2008; Rizos et al., Reference Rizos, Clemente, Bermejo‐Alvarez, de La Fuente, Lonergan and Gutiérrez‐Adán2008; Pontes et al., Reference Pontes, Nonato-Junior, Sanches, Ereno-Junior, Uvo, Barreiros, Oliveira, Hasler and Seneda2009). Numerous studies have mimicked in vivo conditions with the aim of discovering the optimum in vitro embryo culture conditions. The oviduct is a convoluted tube that has an epithelial layer lined with a simple cuboidal-to-columnar epithelium covered by both ciliated and secretory cells (Steinhauer et al., Reference Steinhauer, Boos and Günzel-Apel2004; Ferraz et al., Reference Ferraz, Henning, Stout, Vos and Gadella2017). The oviduct’s secretory epithelial cells produce the proteins and microelements needed for normal gamete and embryo development (Rodriguez-Martinez, Reference Rodriguez-Martinez2007; Mondéjar et al., Reference Mondéjar, Acuna, Izquierdo-Rico, Coy and Avilés2012; Cordova et al., Reference Cordova, Perreau, Uzbekova, Ponsart, Locatelli and Mermillod2014; Schmaltz-Panneau et al., Reference Schmaltz-Panneau, Cordova, Dhorne-Pollet, Hennequet-Antier, Uzbekova, Martinot, Doret, Martin, Mermillod and Locatelli2014). The oviduct also acts as an environment for sperm capacitation (Rodriguez-Martinez, Reference Rodriguez-Martinez2007), sperm storage, acrosome reaction and reducing the possibility of polyspermy (Ghersevich et al., Reference Ghersevich, Massa and Zumoffen2015).
When a follicle ruptures, its cumulus–oocyte complex (COC) ovulates into the infundibulum part of the oviduct. The role of the oviduct in transporting the oocyte to the fertilization site has been attributed to cilia beating and smooth muscle activity, which oestrogen can increase and which progesterone can decrease (Coy et al., Reference Coy, Garcia-Vázquez, Visconti and Avilés2012). When the COC is transported into the ampulla, it remains attached to the epithelium for a variable amount of time depending on the species. Digital video microscope studies have shown that unfertilized porcine and bovine oocytes reached the ampullary–isthmic region within 30–45 min and 3–6 h post ovulation, respectively (Hunter, Reference Hunter1974; Tanghe et al., Reference Tanghe, Van Soom, Nauwynck, Coryn and de Kruif2002; Coy et al., Reference Coy, Garcia-Vázquez, Visconti and Avilés2012). In vitro exposure of oocytes to oviductal cells or oviductal fluid increased sperm–zona binding, zona penetration and fertilization rates in mares, cows and pigs (Mugnier et al., Reference Mugnier, Kervella, Douet, Canepa, Pascal, Deleuze, Duchamp, Monget and Goudet2009; Coy et al., Reference Coy, Lloyd, Romar, Satake, Matás, Gadea and Holt2010). In equine, oocyte exposure to osteopontin, a protein secreted by oviductal epithelium cells, increased in vitro blastocyst yield (Mugnier et al., Reference Mugnier, Kervella, Douet, Canepa, Pascal, Deleuze, Duchamp, Monget and Goudet2009). In mice, pre-treatment of COCs with S100A11, a protein detected at the oestrous stage of the oestrous cycle in the ampullary region of the oviductal epithelium, increased in vitro blastocyst yield (Hanaue et al., Reference Hanaue, Miwa, Uebi, Fukuda, Katagiri and Takamatsu2011). Secretions of different parts of the oviduct at different stages of the oestrous cycle have various effects on gamete function and embryo development (Way et al., Reference Way, Schuler and Killian1997; Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Maillo, Gutierrez-Adán, Bermejo-Alvarez, Ramirez, Yáñez-Mó and Rizos2017). Embryo mRNA synthesis starts in the oviduct at the 8- to 16-cell stage (Graf et al., Reference Graf, Krebs, Heininen-Brown, Zakhartchenko, Blum and Wolf2014). During early embryo development in the oviduct, epigenetic modifications including DNA methylation or histone changes are associated with embryonic implantation, maintenance of pregnancy and the health of the progeny (Fazeli and Pewsey, Reference Fazeli and Pewsey2008; Gad et al., Reference Gad, Hoelker, Besenfelder, Havlicek, Cinar, Rings, Held, Dufort, Sirard and Schellander2012). As in vitro culture conditions have not been completely optimized for normal early embryo development, many researchers have attempted to improve the quality of in vitro embryos by co-culturing gametes and embryos with oviductal epithelial cells (Kattal et al., Reference Kattal, Cohen and Barmat2008; Mugnier et al., Reference Mugnier, Kervella, Douet, Canepa, Pascal, Deleuze, Duchamp, Monget and Goudet2009). Final in vivo maturation of gametes and early embryo development occurs in the oviduct, therefore there is a vital need to obtain deeper knowledge of the oviduct microenvironment that would help researchers and practitioners to improve the efficiency of assisted reproduction techniques (Avilés et al., Reference Avilés, Coy and Rizos2015). Fresh co-culture has been shown to be methodologically complex, lack of reproducibility and have biosanitary risks (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016). One alternative to decrease the variability of co-culture systems could be to use stored frozen oviductal epithelial cells (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016). The aim of this study was to evaluate the effects of different timing of frozen–thawed bovine ampullary epithelial cell (BAECs) and bovine oviductal epithelial cell (BOECs) co-cultures on the development and quality of bovine embryos produced in vitro. Embryo development was assessed as day 8 blastocyst yield, and embryo quality was evaluated as blastocyst differential cell number, cryotolerance and expression of selected genes related to implantation (PLAC8 and IFN-T), apoptosis (BAX and TP53) and cryotolerance (AQP3 and ATP1A1).
Materials and Methods
All the chemicals used in the study were purchased from Sigma (St. Louis, MO, USA) unless otherwise stated.
BOEC isolation and freezing
Oviducts and ovaries from slaughtered cows at the early luteal phase of the oestrus cycle (based on corpus luteum morphology) were transported to the laboratory on ice. The connective tissue around the oviducts was removed, and oviducts were washed twice in normal saline solution and 70% ethanol solution. Then, oviducts were trimmed completely, washed with phosphate-buffered saline (PBS) containing gentamicin and kept in TCM-199 medium (Sigma, m4530) until further utilization. Next, the oviducts were divided into two groups: the first group was kept intact, while in the second group, the ampulla of the oviduct was separated at the ampullary–isthmus junction (identified by where oviduct diameter first exhibited a marked reduction in size). The mucosa of the oviductal region (complete oviduct or the ampulla) was expelled by squeezing with a sterile glass slide. Then, the oviductal and ampullary epithelial cells were collected and transferred to washing medium (TCM-199 supplemented with gentamicin 40 µg/ml; Sigma G1272) and amphotericin B (1 µl/ml; Biowest, L0009-100). Oviductal epithelial cells were washed twice by centrifuging at 400 g for 5 min in the washing medium. Next, trypsin–EDTA enzyme was added to the samples for around 3 min at 37°C to obtain single cells. The enzyme was neutralized using a washing medium supplemented with 10% fetal bovine serum (FBS) (Gibco, 10270). Retrieved fresh epithelial single cells were counted using a haemocytometer and methylene blue staining. The quality of the fresh oviductal epithelial cells was evaluated by seeding 1 × 106 cells/ml in the culture medium (TCM-199, 15% fetal calf serum (FCS) and 40 µg/ml gentamicin). The remaining fresh cells (1 × 107 cell/ml concentration) were frozen in FCS-containing cryoprotectant (10% dimethyl sulfoxide (DMSO)) using a slow freezing procedure described by Freshney (Reference Freshney2015) with slight modifications. Briefly, the cells with cryoprotectant were placed in a −20°C freezer for around 4 h, then transferred to a −80°C freezer for at least one night. The frozen cells were stored in liquid nitrogen until cell culturing. All samples of fresh oviductal cells were cultured to examine the quality of their monolayers. Only those cells with an acceptable quality of monolayer were kept frozen in the cell bank for subsequent oocyte and embryo co-culturing.
BOEC thawing and seeding
At 5 days before using the cell monolayer in the embryo culture, frozen cells obtained from a mixture of oviducts and belonging to one pooled culture were thawed in 37°C sterile water, added to culture medium (TCM-199, 15% FCS and 40 µg/ml gentamicin) and centrifuged at 400 g for 5 min to remove DMSO. Next, the cells were re-suspended in warm culture medium and seeded into four-well NUNC dishes at a concentration of 1 × 106 cell/ml. Medium was refreshed every 48 h during cell culture. At 1 day before co-culture of oocytes or embryos with epithelial cells, the culture medium was replaced with either maturation medium (for co-culture of oocytes with ampullary epithelial cells) or synthetic oviductal fluid (SOF) medium (for co-culture of embryos with whole oviductal epithelial cells).
Comparing the monolayer of fresh and frozen–thawed BOEC
Consistency of growth of the fresh and frozen–thawed BOECs was evaluated by culturing both types of BOECs under similar conditions. Cells were monitored using an inverted phase-contrast light microscope (Hund Wetzlar Wilovert 30, Germany) on days 1, 3, 5 and 7 to ensure that they were healthy and growing as expected.
In vitro embryo production
Oocyte collection and in vitro maturation
Bovine ovaries were transported from the local slaughterhouse (Shiraz, Iran) to the laboratory in saline solution kept at 33–35°C. Immature COCs were collected by aspirating 2–8 mm diameter follicles with a syringe and 18G needle. Class I and class II COCs were selected, washed in warm equilibrated wash medium (HEPES-buffered TCM-199 (M4530), 10% FCS and 50 µg/ml gentamicin (G1272)) three times (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016) and then divided into four groups of 40–45 COCs. COCs were matured in a 500 µl maturation medium with or without bovine ampullary oviductal epithelial cell (BAOEC) monolayers under a 90% humidified atmosphere of 5.5% CO2 in air at 38.5°C in four-well culture dishes (NUNC, Denmark) for 22–24 h. The oocyte maturation medium contained TCM-199 supplemented with 10% FCS, 5 IU/ml of highly purified human chorionic gonadotropin (hCG) (Karma, Pharmatech GmbH, Germany), 0.1 IU/ml of recombinant human follicle-stimulating hormone (FSH) (Follitrope, LG Life Sciences, South Korea) and 10 ng/ml of epidermal growth factor (E4127).
Sperm preparation and in vitro fertilization
Sperm separation and selection was performed using the swim-up procedure described by Brackett and Oliphant (Reference Brackett and Oliphant1975). Briefly, frozen–thawed semen straws from the same bull with proven field fertility were placed at the bottom of a 15-ml Falcon tube and incubated in warm sperm capacitation medium (modified calcium-free Tyrode’s medium supplemented with 1.12 mg/ml Na lactate, 2 mg/ml Na bicarbonate, 0.1 mg/ml caffeine, 1 mg/ml Na pyruvate, 50 µg/ml gentamicin and 6 mg/ml BSA) for 40–50 min. Next, the upper medium was collected and centrifuged at 200 g for 10 min. The sperm pellet was isolated and re-suspended in warm fertilization medium (modified Tyrode’s medium supplemented with 1.12 mg/ml Na lactate, 2.2 mg/ml Na bicarbonate, 0.27 mg/ml caffeine, 1 mg/ml Na pyruvate, 10 µg/µl heparin, 50 µg/µl hypotaurine, 50 µg/ml gentamicin and 6 mg/ml BSA). Matured COCs were washed three times in fertilization medium and transferred into four-well dishes containing 450 µl of IVF medium. Sperm concentration was determined, and a concentration of 1 × 106 sperm/ml was used for in vitro insemination (Azari et al., Reference Azari, Kafi, Ebrahimi, Fatehi and Jamalzadeh2017).
In vitro culture of the presumptive zygote
Presumptive zygotes were denuded around 18–20 h post insemination (p.i.) by repeated pipetting. Cumulus cells and excess spermatozoa were deleted by washing in modified SOF (mSOF). Next, the cleaned zygotes were placed into four-well dishes (NUNC, Denmark) containing SOF medium supplemented with 5% bovine calf serum (BCS), 4 mg/ml fatty acid-free BSA (Sigma, A3675) and 50 µg/ml gentamicin with or without a BOECs monolayer. The culture medium was refreshed every 48 h, and cultures were incubated under 90% humidified conditions at 38.5°C in an atmosphere of 5% CO2 in air.
Assessment of embryo development and quality
Embryo development
Embryo development was determined by cleavage rate (defined as the total number of cleaved zygotes/total number of fertilized oocytes) and blastocyst yield (defined as the total number of blastocysts/total number of cleaved zygotes). Cleavage rate was recorded on day 2 (48 h p.i.), and the blastocyst yield was recorded on days 7 and 8 (p.i. under a stereomicroscope).
Embryo quality
Quality of the expanded blastocysts at days 7 and 8 p.i was evaluated by cryotolerance assessment, differential cell count and expression analysis of the selected genes.
Blastocyst vitrification. Blastocyst cryotolerance was assessed by vitrification-warming of early- and mid-stage blastocysts using the procedure described by Rizos et al. (Reference Rizos, Ward, Duffy, Boland and Lonergan2002). Blastocyst vitrification was performed in two steps: first, the blastocysts were washed in a holding medium (HM) (TCM-199–M4530 supplemented with 20% (v/v) FCS). Blastocysts were then transferred to HM1 medium (HM containing 7.5% ethylene glycol and 7.5% DMSO) for about 3 min. Second, the blastocysts were transferred to HM2 medium (HM supplemented with 16.5% ethylene glycol, 16.5% DMSO and 0.5 M sucrose) for up to 30 s. Then, blastocysts were placed on a cryotop and quickly floated in liquid nitrogen. In the thawing process, the vitrified blastocysts were warmed twice, initially in HM medium (supplemented with 0.25 M sucrose) and then in HM medium (supplemented with 0.15 M sucrose). Next, blastocysts were cultured in SOF medium with 5% FCS in four-well NUNC dishes. The survival rate of the vitrified-warmed blastocysts was investigated as the re-expansion of the blastocoel and its maintenance for 4 h, 24 h and 48 h post culture.
Differential staining of blastocysts. Differential staining of the inner cell mass (ICM) and trophectoderm (TE) was performed using the procedure described by Thouas et al. (Reference Thouas, Korfiatis, French, Jones and Trounson2001) with slight modifications. Briefly, the expanded blastocysts were incubated in solution I (5 mg/ml BSA–TCM-199 with 1% Triton X-100 (T8787) and 100 µg/ml propidium iodide (P4170)) for up to 30 s in the dark to stain the TE. Next, blastocysts were immediately transferred to solution II (100% ethanol with 25 µg/ml bisbenzimide H33342 (B2261)) and kept for 30 min in the dark to complete the fixation and the ICM staining processes. Stained blastocysts were transferred to a glycerol droplet and mounted onto a glass microscope slide. Cell counting was carried out using a fluorescence microscope (Olympus BX61, Japan).
Gene expression analysis. Expression levels for six target genes (two apoptosis-related genes (BAX and TP53), two cryotolerance-related genes (AQP3 and ATP1A1) and two implantation-related genes (PLAC8 and IFN-T)) along with two housekeeping genes (reference genes (GAPDH and YWHAZ)) were assessed using quantitative RT-PCR (qPCR). The names, primers (designed using PerlPrimer software; Marshall, Reference Marshall2004), GenBank accession numbers and the expected product sizes of the genes are presented in Table 1. Total RNA was isolated using an RNeasy Micro Kit (catalogue no. 74004, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Total RNA concentrations were determined using a NanoDrop 2000 spectrophotometer (absorbance at 260 nm wavelength). All samples were treated with DNase and RQ1 DNase (Promega, France) to remove possible DNA contamination. Sample purity was assessed based on the A260/A280 nm ratio, with ratios expected to fall between 1.8 and 2.0. Immediately after RNA extraction, reverse-transcription (RT) was performed using a RevertAid H Minus First Strand cDNA Synthesis Kit (K1632, Fermentas, Germany). All mRNA transcripts for the mentioned genes were quantified using qPCR, with three replicates for each group. The PCR mix in each well contained 5 μl of Power SYBR® Green PCR Master Mix (Applied Biosystems, UK), 1 μl of 5 pM forward and reverse primers, 2 μl of cDNA (12.5 ng/ μl) and 11 μl dH2O in a final volume of 20 ml. PCR reactions were carried out in the Applied Biosystems StepOne and StepOnePlus Real-Time PCR system using the following program: Stage 1: 95°C for 10 min. Stage 2: 95°C for 15 s and 60°C for 1 min for in total, 40 cycles. Stage 3: 95°C for 15 s, 60°C for 15 s and 95°C for 15 s. The last heating step in Stage 3 was performed with a ramp rate of 2% to enable the generation of a dissociation curve of the products. Each reaction was run in duplicate, and the mean value of each duplicate was used for the further calculations. The expression data were normalized using the geometric mean of the internal reference genes (GAPDH and YWHAZ). Then, 2–ΔCT (geometric mean of housekeeping genes) values were calculated for each gene.
Table 1. Details of primers used for qRT-PCR

Experimental design
A schematic diagram of the experimental design for this study is shown in Fig. 1. The study was carried out using the treatments described below.

Figure 1. Control: IVM, IVF, and IVC was performed using a cell-free culture medium. AM: IVM was performed using TCM-199 for 18 h and then transferred into a medium containing a monolayer of frozen–thawed bovine ampullary cells for the remaining 6 h of culture. OV: the presumptive zygotes were co-cultured using a monolayer of bovine frozen–thawed oviductal cells for 4 days and then with a cell-free medium for the last 4 days of culture. AMOV: oocytes were cultured similarly to those in the AM group, and the presumptive zygotes were co-cultured similarly to those in the OV group.
Control group
The oocytes were placed into cell-free maturation medium for 24 h (IVM). Next, the matured oocytes were transferred into dishes containing IVF medium and capacitated sperms. At 18–20 h p.i, the presumptive zygotes were denuded and transferred to the plates containing SOF medium for 8 days consecutively (IVC).
AM group
Oocytes were cultured in cell-free maturation medium for 18 h. For the last 6 h in vitro maturation they were transferred to medium over a BAOECs monolayer (mimicking the in vivo ampulla conditions). IVF and IVC steps were performed as described for the control group.
OV group
IVM and IVF steps were performed as described for the control group. The denuded presumptive zygotes were placed in NUNC plates containing a bovine total oviductal epithelial cell (BTOECs) monolayer for 4 days p.i. Then, embryos were transferred to wells containing cell-free medium for the last 4 days of culture (days 5–8 p.i.).
AMOV group
Oocytes were cultured in maturation medium for 18 h and then transferred to medium containing a BAECs monolayer for the last 6 h of in vitro maturation. Next, the matured oocytes were transferred to dishes containing in vitro fertilization medium and the capacitated sperms. The presumptive zygotes were denuded and placed into four-well NUNC dishes containing a BTOEC monolayer for 4 days p.i. and transferred into wells containing cell-free medium for the last 4 days of culture (days 5–8 p.i.). The culture media were refreshed every 48 h for all treatments.
The overall cleavage rate was recorded at 48 h p.i.; blastocyst development was recorded on days 7 and 8 p.i. A representative number of day 7–8 blastocysts from each group (treatment) were either vitrified-warmed for the survival rate analysis at 4 h, 24 h and 48 h post warming, fixed for the differential cell count or frozen in liquid nitrogen and stored at −80°C for gene expression analysis. Eight replicates were generated for each treatment.
Statistical analysis
Data on the cleavage rate, blastocyst yield, survival rate of the vitrified-warmed blastocysts, embryo cell number (ICM, TE, ICM/TE ratio) and relative mRNA abundance were analyzed using one-way analysis of variance (ANOVA) in R (R Core Team, 2017). The assumptions of normality and homogeneity of the residual variances were checked at P-value < 0.05 using Shapiro–Wilk and Bartlett tests, respectively. Differences were considered to be statistically significant at P < 0.05.
Results
Comparing the monolayer of fresh versus frozen–thawed BOECs
Fresh and frozen–thawed BOEC monolayers showed similar morphology and growing patterns on days 3 and 5 of culture (Fig. 2).
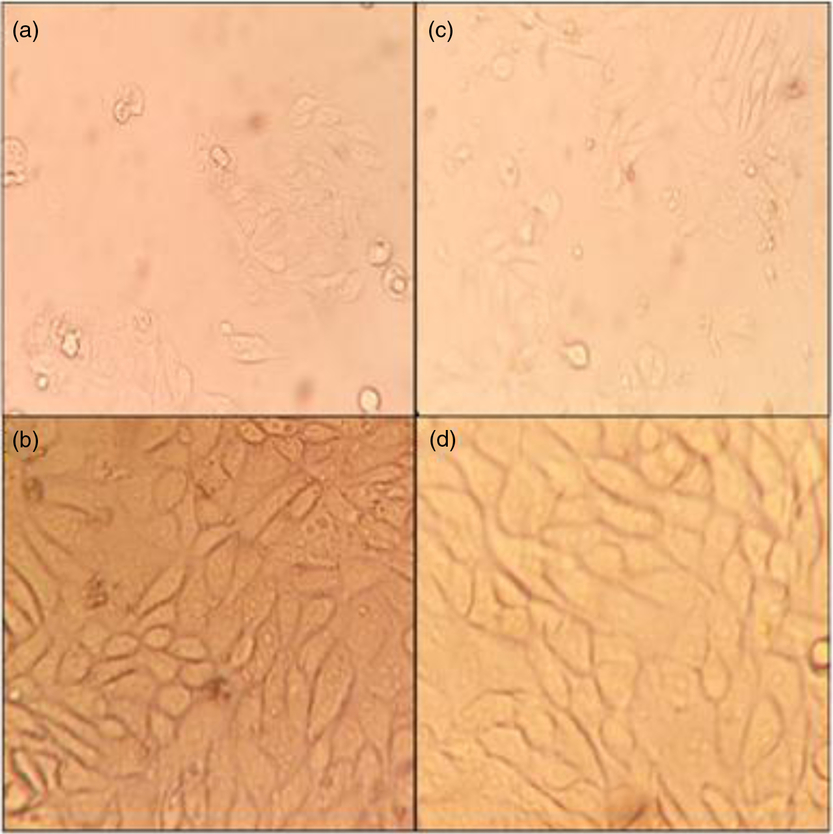
Figure 2. BOEC morphology. Fresh BOECs monolayer on day 3 (a) and day 5 (b). Frozen–thawed BOECs monolayer on day 3 (c) and day 5 (d); ×40 magnification.
Embryo development
Effects of co-culture conditions on the cleavage rate and blastocyst yield are presented in Table 2. Although various co-culture conditions had no effect on cleavage rate (P ≥ 0.05), blastocyst yield was affected by the treatments (P < 0.05). The highest blastocyst yield was found in the AM group, followed by the control group. However, no difference was found between the OV and AMOV groups (Table 2).
Table 2. Effect of the experimental treatments on the in vitro embryo development (n = 8 replicates)

1 Control: IVM, IVF, and IVC was performed using a cell-free culture medium. AM: IVM was performed using TCM-199 for 18 h and then transferred into a medium containing a monolayer of frozen–thawed bovine ampullary cells for the remaining 6 h of culture. OV: the presumptive zygotes were co-cultured using a monolayer of bovine frozen–thawed oviductal cells for 4 days and then with a cell-free medium for the last 4 days of culture. AMOV: oocytes were cultured similarly to those in the AM group, and the presumptive zygotes were co-cultured similarly to those in the OV group (mean ± standard error of the mean (SEM)).
2 Number of cleaved zygotes on day 2 out of total number of fertilized oocytes.
3 Number of blastocysts (day 8) out of number of cleaved zygotes.
a,b,c Values in the same column with different superscripts differ significantly (P < 0.05).
Blastocyst cryotolerance
The effects of co-culture conditions on the survival and hatching rates of vitrified-warmed blastocysts are presented in Table 3. The 4 h post-warming survival of vitrified-warmed blastocysts was not affected by the co-culture conditions (P ≥ 0.05), whereas co-culture conditions for the AM and AMOV groups resulted in a higher survival rate at 24 and 48 h post warming, as well as a higher 48 h post-warming hatching rate than for the OV and control treatments (P < 0.05).
Table 3. Effect of experimental treatments on the survival and 48-h post-culture hatch rate of the vitrified-warmed day 7–8 blastocysts

1 Control: IVM, IVF, and IVC was performed using a cell-free culture medium. AM: IVM was performed using TCM-199 for 18 h and then transferred into a medium containing a monolayer of frozen–thawed bovine ampullary cells for the remaining 6 h of culture. OV: the presumptive zygotes were co-cultured using a monolayer of bovine frozen–thawed oviductal cells for 4 days and then with a cell-free medium for the last 4 days of culture. AMOV: oocytes were cultured similarly to those in the AM group, and the presumptive zygotes were co-cultured similarly to those in the OV group (mean ± standard error of the mean (SEM)).
2 Total number of vitrified-warmed day 8 blastocysts.
a,b Values in the same column with different superscripts differ significantly (P < 0.05).
Differential cell count
The effects of experimental treatments on differential cell counts are presented in Table 4. The lowest average number of ICM, TE and total blastomere cells was found in the control group, whereas there was no difference in cell count when using various co-culture conditions in the AM, AMOV and OV groups (Table 4).
Table 4. Effect of the experimental treatments on the differential cell count measures of day 8 blastocysts

1 Control: IVM, IVF, and IVC was performed using a cell-free culture medium. AM: IVM was performed using TCM-199 for 18 h and then transferred into a medium containing a monolayer of frozen–thawed bovine ampullary cells for the remaining 6 h of culture. OV: the presumptive zygotes were co-cultured using a monolayer of bovine frozen–thawed oviductal cells for 4 days and then with a cell-free medium for the last 4 days of culture. AMOV: oocytes were cultured similarly to those in the AM group, and the presumptive zygotes were co-cultured similarly to those in the OV group (mean ± standard error of the mean (SEM)).
2 Total number of day 8 blastocysts fixed for the differential cell count.
3 Inner cell mass.
4 Trophectoderm.
5 Inner cell mass plus trophectoderm.
a,b Values in the same column with different superscripts differ significantly (P < 0.05).
Gene expression analysis
Effects of the experimental treatments on the expression levels for six target genes (two apoptosis-related genes (BAX and TP53), two cryotolerance-related genes (AQP3 and ATP1A1) and two implantation-related genes (PLAC8 and IFN-T)) are presented in Fig. 3. BAX was significantly downregulated in the OV group compared with the rest of the groups (P < 0.05). The P53 gene showed no difference in expression among groups (P ≥ 0.05). Expression of the IFN-T gene was downregulated in all co-culture conditions compared with the control group; its relative expression in the OV group was lower than that in the AM and AMOV groups (P < 0.05). Highest expression levels for PLAC8 and AQP3 were found in the AMOV group, followed by the AM group; there was no difference in expression between the control and OV groups (P ≥ 0.05). The ATP1A1 gene showed the highest expression level for samples in the AMOV group, followed by the OV group; there was no difference between expression levels in the control and AM groups (P ≥ 0.05).

Figure 3. Relative mRNA expression levels from apoptosis genes (BAX and P53), implantation genes (INF-T and PLAC8), and cryotolerance genes (ATP1A1 and AQP3) in day 8 blastocysts cultured in different co-culturing treatments. Data are relative to the geometric mean of internal genes GAPDH and YWHAZ. a,b,cDifferent letters indicate significant differences for each gene (P < 0.05). Control: IVM, IVF, and IVC was performed using cell-free culture medium. AM: IVM was performed using TCM-199 for 18 h and then transferred into a medium containing monolayer of frozen–thawed bovine ampullary cells for the remaining 6 h of culture. OV: the presumptive zygotes were co-cultured using monolayer of bovine frozen–thawed oviductal cells for 4 days and then with cell-free medium for the last 4 days of culture. AMOV: The oocytes were cultured similarly to those in the AM group, and the presumptive zygotes were co-cultured similarly to those in the OV group (mean ± standard error of the mean (SEM)).
Discussion
Fresh co-culture is associated with methodological complexity, lack of reproducibility and biosanitary risks (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016). This study optimized a procedure to provide a cell bank of fresh BOECs. In this procedure, fresh bovine oviductal epithelial single cells were frozen and successfully thawed, a process that can improve the reproducibility and time efficiency of cell culture. Variation may exist among fresh BOEC lines recovered from the oviduct of different cows. However, the use of frozen–thawed BOECs makes it possible to find the most appropriate cell line for BOECs and use it to consistently produce bovine blastocysts with less laboratory complications and in a more predictable co-culture system. In addition, using the same pooled frozen–thawed oviductal epithelial cells and different experimental replicates can increase reproducibility. To the best of our knowledge, this study is the first to document that a monolayer of frozen–thawed BAECs can be used as an alternative method for in vitro maturation of bovine oocytes.
Although there has been considerable success in increasing the efficiency of IVP, the quality of the produced embryos still remains lower than that of embryos produced in vivo (Lonergan and Fair, Reference Lonergan and Fair2008; Rizos et al., Reference Rizos, Clemente, Bermejo‐Alvarez, de La Fuente, Lonergan and Gutiérrez‐Adán2008; Pontes et al., Reference Pontes, Nonato-Junior, Sanches, Ereno-Junior, Uvo, Barreiros, Oliveira, Hasler and Seneda2009). This situation illustrates that researchers must attempt to create an in vitro environment that is relatively similar to conditions during final maturation of the oocyte in the oviduct. Evidence suggests that the quality of the oocyte at the start of IVM is a vital determinant of the proportion of oocytes that will reach the blastocyst stage (Lonergan and Fair, Reference Lonergan and Fair2016). By mimicking in vivo conditions, a better quality of matured oocyte can be obtained, and this can have a positive effect on developmental potential of early embryos throughout IVP (Avilés et al., Reference Avilés, Coy and Rizos2015). Under in vivo conditions, bovine COCs are situated in the ampulla part of the oviduct for the first few hours after ovulation to complete the maturation process. The oviduct provides nutrients and enzymes that have anti-oxidant effects, and that are useful for oocyte survival (Leese et al., Reference Leese, Hugentobler, Gray, Morris, Sturmey, Whitear and Sreenan2007; Avilés et al., Reference Avilés, Gutierrez-Adán and Coy2010). The main aim of this study was to mimic these in vivo conditions to improve the COC maturation process in an IVM procedure. The most important finding of this study was that blastocyst yield and the quality of the produced blastocysts improved when the last 6 h of in vitro oocyte maturation was carried out in the presence of a frozen–thawed BAOECs monolayer. Previous researchers have reported that the presence of oviductal cells or fluid from cows in the non-luteal phase of the oestrous cycle had a positive effect on in vitro sperm–egg binding and fertilization rates (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Maillo, Gutierrez-Adán, Bermejo-Alvarez, Ramirez, Yáñez-Mó and Rizos2017). When the oocyte moves forward in the ampulla, it becomes exposed to different proteins and factors such as oviduct-specific glycoprotein 1 and oviductine, which are needed for sperm–zona binding and zona hardening, respectively (Coy et al., Reference Coy, Cánovas, Mondéjar, Saavedra, Romar, Grullón, Matás and Avilés2008; Ghersevich et al., Reference Ghersevich, Massa and Zumoffen2015). When the COC moves forward in the ampulla, it is influenced continuously by factors secreted from the ampulla epithelial cells that are vital for the improvement of sperm–zona binding, zona pellucida (ZP) hardening and fertilization rate (Coy et al., Reference Coy, Cánovas, Mondéjar, Saavedra, Romar, Grullón, Matás and Avilés2008; Mugnier et al., Reference Mugnier, Kervella, Douet, Canepa, Pascal, Deleuze, Duchamp, Monget and Goudet2009; Hanaue et al., Reference Hanaue, Miwa, Uebi, Fukuda, Katagiri and Takamatsu2011; Coy et al., Reference Coy, Garcia-Vázquez, Visconti and Avilés2012; Ghersevich et al., Reference Ghersevich, Massa and Zumoffen2015). There was a higher blastocyst yield in the AM group than in the AMOV group, in which monolayers of frozen/thawed BAOECs and BTOECs were used for oocyte maturation and embryo culture, respectively. This finding could be partly explained by the fact that under in vivo conditions, continuous renewal of oviductal fluid creates an optimal microenvironment for embryo development, whereas the accumulation of waste metabolites during co-culture adversely affects early-stage embryo development (Buhi, Reference Buhi2002).
The blastocyst’s ability to withstand cryopreservation can be used as an indicator for assessing embryo quality (Rizos et al. Reference Rizos, Ward, Duffy, Boland and Lonergan2002, Reference Rizos, Clemente, Bermejo‐Alvarez, de La Fuente, Lonergan and Gutiérrez‐Adán2008; Moore et al., Reference Moore, Rodríguez-Sallaberry, Kramer, Johnson, Wroclawska, Goicoa and Niasari-Naslaji2007). In the present study, cryotolerance at 4 h post thawing was not affected by the co-culture conditions. However, blastocysts in the AM and AMOV groups showed higher cryotolerance at both 24 and 48 h post thawing compared with the OV and control groups. Previous studies have shown that using BOEC (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016) and oviductal fluid (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Maillo, Gutierrez-Adán, Bermejo-Alvarez, Ramirez, Yáñez-Mó and Rizos2017) in in vitro embryo cultures improved the survival rate of vitrified-warmed blastocysts.
It is well documented that TE cells play a crucial role in implantation and placentation (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016). The present results showed that the numbers of ICM and TE cells present in all co-culture treatments were higher compared with the numbers in the control group; this finding was in close agreement with results of previous studies (Galli and Lazzari, Reference Galli and Lazzari1996; Rizos et al., Reference Rizos, Ward, Boland and Lonergan2001; Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016). These findings can be partly explained because TE cells are essential for blastocyst re-expansion and preservation after vitrification (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Fernandez-Fuertes, Maillo, Beltrán-Breña, Calle, Redruello, López-Martín, Gutierrez-Adán and Yáñez-Mó2016).
Expression patterns of relevant genes are a useful tool for tracking and predicting the clinical outcome of IVF (Lonergan et al., Reference Lonergan, Rizos, Kanka, Nemcova, Mbaye, Kingston, Wade, Duffy and Boland2003; Jurisicova et al., Reference Jurisicova, Antenos, Varmuza, Tilly and Casper2003; Corcoran et al., Reference Corcoran, Rizos, Fair, Evans and Lonergan2007). This study showed that the expression patterns of genes – including BAX, INF-T, PLAC8, ATP1A1 and AQP3 – in day 8 blastocysts were influenced by co-culture conditions. However, the P53 expression level was not affected. Na/K-ATPase α-1 (ATP1A1) is a gene involved in membrane trafficking (Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Maillo, Gutierrez-Adán, Bermejo-Alvarez, Ramirez, Yáñez-Mó and Rizos2017), and its product is essential for blastocyst expansion (Barcroft et al., Reference Barcroft, Moseley, Lingrel and Watson2004). Monitoring its expression is a suitable way to assess cryotolerance. The highest expression level for ATP1A1 was found in the AMOV group and the lowest levels were found in the control and AM groups. Hosseini et al. (Reference Hosseini, Asgari, Ostadhosseini, Hajian, Ghanaei and Nasr-Esfahani2015) reported that the number of ATP1A1 transcripts in high-quality ovine embryos was significantly higher than in those groups with poor-quality embryos. The higher survival rate of the vitrified-warmed blastocysts in the AMOV group could be partly explained by the higher level of ATP1A1 expression in this group. The highest expression level for AQP3 was found in the AMOV group, followed by the AM group. One of the well known molecular explanations for assessing cryotolerance ability after vitrification is the water channel aquaporin (AQP3) protein, which is involved in membrane permeability regulation (Cordova et al., Reference Cordova, Perreau, Uzbekova, Ponsart, Locatelli and Mermillod2014). This gene codes for a protein involved in facilitating rapid water movement through the membrane and therefore increases the survival rate of the vitrified-warmed blastocysts (Huang et al., Reference Huang, He, Sun, Zhang, Meng and Ma2006; Lopera-Vásquez et al., Reference Lopera-Vásquez, Hamdi, Maillo, Gutierrez-Adán, Bermejo-Alvarez, Ramirez, Yáñez-Mó and Rizos2017). Consequently, upregulation of the AQP3 gene increases the survival rate and dummy expression of AQP3 in mouse oocytes increased cryotolerance (Edashige et al., Reference Edashige, Yamaji, Kleinhans and Kasai2003). In this study, results for the obtained cryotolerance data correlated with the observed expression level for AQP3 in the treatments. Cordova et al. (Reference Cordova, Perreau, Uzbekova, Ponsart, Locatelli and Mermillod2014) reported that there was no difference between the expression levels for AQP3 in the groups co-cultured with BOEC in the first 4 days of IVC compared with the expression level in the control group.
PLAC8 is related to successful embryo development, more particularly it plays a role in feto-maternal communication and placental development (Galaviz-Hernandez et al., Reference Galaviz-Hernandez, Stagg, De Ridder, Tanaka, Ko, Schlessinger and Nagaraja2003). PLAC8 and AQP3 showed similar expression patterns under different treatments. IFN-T gene products modify ovarian progesterone secretion and stimulate endometrial nutrient secretion, which is required for growth of the conceptus. In addition, IFN-T gene products regulate the expression of uterine-derived factors, which are responsible for the early development of the conceptus and placental attachment (Austin et al., Reference Austin, Ward, Glaucia Teixeira, Dean, Moore and Hansen1996). In this study, the lowest expression level for IFN-T was found in the OV group; this finding partly explains the lowest rate of development of blastocysts in this group.
Apoptosis, a complex programmed cell death process that is activated in embryonic cells in stressful conditions, can be evaluated by examining the expression patterns of the involved genes (e.g. BAX) (Betts and King, Reference Betts and King2001). BAX is a proapoptotic gene (Stallock et al., Reference Stallock, Molyneaux, Schaible, Knudson and Wylie2003) and can be used as a potential marker for embryo quality. Melka et al. (Reference Melka, Rings, Hölker, Tholen, Havlicek, Besenfelder, Schellander and Tesfaye2010) showed that the expression level of BAX in poor-quality embryos was higher than that in high-quality embryos. The present results showed that the expression level of the BAX gene in the OV group was lower than that in the other groups. However, the pattern of expression of BAX among groups did not explain the observed survival rate of the vitrified-warmed blastocysts. The P53 gene, also known as TP53, codes for a protein that regulates the cell cycle. P53 plays an important role in conserving stability by preventing genome mutation (Strachan and Read, Reference Strachan and Read2018). However, the present results showed that the treatments did not affect P53 expression. Similarly, Cordova et al. (Reference Cordova, Perreau, Uzbekova, Ponsart, Locatelli and Mermillod2014) reported no difference between expression levels of P53 in the control and in the BOEC co-culture treatments.
Conclusions
This study found that co-culture of bovine oocytes with a monolayer of frozen–thawed bovine ampullary cells during the last 6 h of IVM increased both the blastocyst yield and quality of the produced embryos. The achieved improvement in blastocyst development and embryo quality suggested that using BAOEC co-culture in in vitro maturation provides the conditions needed for oocyte maturation and embryo development. Furthermore, bio-banking of fresh oviductal single cells can provide a reliable and reproducible way for IVM/IVF/IVC to effectively mimic in vivo conditions.
Acknowledgements
We specifically thank Sheryl Nikpoor, the editor of scientific manuscripts at the Shiraz University, for her meticulous editing of our manuscript. The authors also thank A. Shams, the Chief Director of the Shiraz Abattoir for his kind assistance in collecting the ovaries. Thanks also to Z. Heidary of Royan Institute and H. Jesmani for their technical help in preparing the IVF laboratory and performing RT-PCR.
Financial support
This study was financially supported by Shiraz University (grant no. 97GCU1M1251) and the Centre of Excellence for Research on High Producing Dairy Cows.
Conflicts of interest
The authors declare no conflict of interest.
Ethical standards
The ethical and research committee of the School of Veterinary Medicine, Shiraz University (97GCU1M1251) approved this study.







