


INTRODUCTION
The parasite Toxoplasma gondii is a member of the eukaryotic phylum Apicomplexa. The phylum, which includes Plasmodium falciparum, a causative agent of malaria, as well as multiple other parasitic genera, is of immense medical and veterinary importance. Members of this phylum share a number of structural attributes. With the likely exception of Cryptosporidium, one such shared characteristic is the apicoplast, a relict plastid believed to have been acquired through secondary endosymbiosis (Wilson et al. Reference Wilson, Williamson and Preiser1994; McFadden et al. Reference McFadden, Reith, Munholland and Lang-Unnasch1996). The T. gondii apicoplast contains its own ~35 kb genome that is replicated and segregated with the organelle during parasite proliferation. With the exception of the microgamete stage, all T. gondii life-cycle stages have been demonstrated to possess a single apicoplast, which is faithfully segregated into the daughter cells (Striepen et al. Reference Striepen, Crawford, Shaw, Tilney, Seeber and Roos2000; Ferguson et al. Reference Ferguson, Campbell, Henriquez, Phan, Mui, Richards, Muench, Allary, Lu, Prigge, Tomley, Shirley, Rice, McLeod and Roberts2007). Like many endosymbiont-derived organelles, the apicoplast has a reduced genome containing genes involved principally in its own replication. A variety of the genes originally associated with the prokaryotic endosymbiont have been incorporated into the nuclear genome and their products are targeted back to the apicoplast using a bi-partite plastid targeting peptide. Consequently, many nuclear-encoded enzymes associated with the apicoplast are of prokaryotic ancestry. The organelle is essential as it is the site for numerous biochemical pathways including heme and fatty acid biosynthesis and steroid production (Roos, Reference Roos1999; Soldati, Reference Soldati1999; McLeod et al. Reference McLeod, Muench, Rafferty, Kyle, Mui, Kirisits, Mack, Roberts, Samuel, Lyons, Dorris, Milhous and Rice2001). The apicoplast has therefore proven to be a valuable target for drugs active against apicomplexan parasites (Fichera and Roos, Reference Fichera and Roos1997).
Genes encoding proteins involved in DNA replication or repair are absent from the apicoplast genome. Certain genes encoding plastid DNA replication enzymes are, however, present in the nuclear genome, with the protein being imported into the plastid post-translationally. Inhibition of components of the apicoplast DNA replication machinery (e.g. DNA gyrase) by ciprofloxacin (Dar et al. Reference Dar, Sharma, Mondal and Dhar2007) can cause cessation of apicoplast replication and subsequent parasite death (Dar et al. Reference Dar, Sharma, Mondal and Dhar2007; Raghu Ram et al. Reference Raghu Ram, Kumar, Biswas, Kumar, Chaubey, Siddiqi and Habib2007). Thus, the nuclear encoded apicoplast replication machinery is an established drug target in apicoplexan parasites (Ralph et al. Reference Ralph, D'Ombrain and McFadden2001).
The various enzymatic activities required for replication of the apicoplast genome in P. falciparum may be derived in large part from a single gene, pfprex, which encodes a multifunctional protein (Seow et al. Reference Seow, Sato, Janssen, Riehle, Mukhopadhyay, Phillips, Wilson and Barrett2005) comprising a T7 bacteriophage homologous primase-helicase domain and a prokaryotic DNA polymerase domain separated by a spacer domain. An N-terminal sequence of PfPREX targeted Green Fluorescent Protein (GFP) to the apicoplast, strongly suggesting that this protein may be involved in DNA replication or repair of the organelle genome. Thus, this single gene provides multiple functional properties required for apicoplast replication and/or repair (Seow et al. Reference Seow, Sato, Janssen, Riehle, Mukhopadhyay, Phillips, Wilson and Barrett2005). Herein, we demonstrate the presence of a single prex gene (Tgprex) in T. gondii. Unlike prex in P. falciparum, the gene in T. gondii is interrupted by multiple introns. The polymerase domain expressed as a recombinant protein in E. coli displays DNA polymerase activity. The conservation of unusual prokaryotic-like DNA replication enzymes across the Apicomplexa indicates that PREX could be a potential target for chemotherapy against this class of organisms.
MATERIALS AND METHODS
T. gondii tachyzoite preparation and preparation of cDNA
T. gondii tachyzoites (RH strain) were grown in the peritoneum of BALB/c mice as previously described (Roberts and Alexander, Reference Roberts and Alexander1992). Tachyzoites were harvested and washed in sterile phosphate-buffered saline (PBS; 0·1 m NaCl, 2·7 mm KCl, 10 mm Na2HPO4, 1·76 mm KH2PO4, pH 7·4) by centrifugation at 500 g for 5 min at 4°C and stored as pellets at −70°C. RNA isolation from the tachyzoites was performed using Trizol® (Invitrogen, UK) following a protocol, based on a single-step acid guanidinium thiocyanate-phenol-chloroform for RNA isolation (Chomczynski and Sacchi, Reference Chomczynski and Sacchi1987).
Complementary DNA (cDNA) was produced from total RNA as previously described (Campbell et al. Reference Campbell, Richards, Mui, Samuel, Coggins, McLeod and Roberts2004). In a 13 μl volume, 1 μg of RNA was added to 1 μl of random hexamer mix (Promega, Southampton, UK) and 1 μl of a 10 mm deoxynucleoside triphosphate mixture, incubated at 65°C for 5 min, and chilled on ice for 1 min. Then 80 units of RNAsin ribonuclease inhibitor (Promega, UK), 4 μl of 5× first strand buffer (250 mm Tris-HCl, pH 8·3, 375 mm KCl, 15 mm MgCl2), 1 μl of 0·1 m DTT and 200 units of Superscript III reverse transcriptase RNAse H− (Invitrogen, UK) were added to the reaction and incubated for 5 min at 25°C followed by 55°C for 60 min. Inactivation of the reverse transcriptase was performed by heating at 70°C for 10 min.
Identification and sequencing of the prex gene in T. gondii
Initially the PfPREX homologues were identified in the T. gondii genome project by BLASTP analysis. As the P. falciparum gene was AT rich, the protein sequence was used as a query and the protein match was extrapolated to its cognate gene in the genome. The two protein coding genes are situated on chromosome VIIb in the T. gondii genome project (www.toxodb.org release 4) (Gajria et al. Reference Gajria, Bahl, Brestelli, Dommer, Fischer, Gao, Heiges, Iodice, Kissinger, Mackey, Pinney, Roos, Stoeckert, Wang and Brunk2008). Oligonucleotide primers were designed to confirm the prex gene structure by PCR. The primer locations are listed in relation to the actual gene model (Fig. 1A) determined in this study and are listed in order of their positions on the gene (Table 1). Primer pairs for each PCR are designated in Fig. 1A. The primers situated within the consensus exons predicted by different gene prediction algorithms (GLEAN, GlimmerHMM, TwinScan, TwinScan/Eimeria and TigrScan) are in bold font (Table 1). After confirmation of predicted sequences these primers were coupled with primers from other predicted exons to confirm the coding sequence.

Fig. 1. The exon–intron structure (Guo et al. Reference Guo, Zhu, Chen and Luo2007) and a diagrammatic representation of the domain structure of the Toxoplasma gondii ORF 7740 present on chromosome VIIb of Toxoplasma gondii as elucidated by PCR. The structure of the T. gondii ORF 7740 was confirmed by the sequencing of cDNA from the RH strain of the organism, using primers listed in Table 1 (A). The 7740 bp long ORF was composed of 20 exons separated by 19 introns and different reading frames for the introns are designated. The exon intron structure of the gene was created using GSDS (http://gsds.cbi.pku.edu.cn/) (Guo et al. Reference Guo, Zhu, Chen and Luo2007). Primer pairs used for PCR are delineated in relation to the exon structure of the Tgprex gene. The primers designed on the consensus exon sequences are in bold font. (B) 5′RACE was performed to confirm the initiation codon where RT+ and RT− refers to reactions with or without reverse transcriptase, respectively. (C) The 5′ end of the ORF was confirmed by comparing PCR on cDNA (c) and genomic DNA (g) using primers F.I and R3.1 (Table 1) generating 2345 bp and 3178 bp long PCR products, respectively. (D) The continuity of the single gene was confirmed by PCR using primers F11.2 and R12.3 (Table 1) showing a 1766 bp long PCR product from cDNA (c) compared to a 3334 bp long product from genomic DNA (g). The DNA marker is designated by M. (E) The domain structure of the TgPREX putative protein is diagrammatically represented.
Table 1. List of primers used for confirmation of the Tgprex gene coding sequence on chromosome VIIb of Toxoplasma gondii and expression of recombinant polymerase domain of TgPREX protein
(The table shows the primer name, nucleotide sequence and the position of each primer according to the sequenced gene model presented in Fig. 1A. Primer pairs used for PCR amplification are delineated in Fig. 1A. The reverse primers are highlighted in the grey background. Primers situated in consensus exons predicted by ToxoDB are in bold font. The 5′RACE primers were used for the clarification of the initiation codon of the gene that was confirmed by PCR amplification of the 5′ region using the 5′UTR primer. The cDNA primers were used to establish the coding sequence of the gene. The 3′UTR primer was used to ensure the 3′ end of the Tgprex gene. Polymerase domain of the TgPREX protein was cloned using forward primer Tg_polymeraseF in conjunction with R20.2 reverse primer. The Nhe I restriction site on Tg_polymeraseF primer is shown in italicized font.)

PCR reactions were performed on both genomic and cDNA from T. gondii (RH strain) tachyzoites for comparison. One μl of the cDNA or 500 ng of the genomic DNA template was amplified by Expand Hi-Fidelity polymerase enzyme (Roche) using 0·2 mm dNTP mix, 0·4 μm of sequence specific forward and reverse primers with 1·5 μm/2·0 μm of Mg2+ in enzyme-specific buffer. Amplified PCR products from the cDNA were cloned into the pGEM-T Easy vector (Promega) and sequenced to verify the ORF. The cDNA contig was assembled using the Contig Express programme of the Vector NTI suite 9 and 10.
To clarify the 5′ end of the coding sequence in the prex cDNA, Rapid Amplification of the cDNA end (RACE) was performed on T. gondii total RNA. The cDNA was amplified (Superscript III, Invitrogen) using a gene-specific reverse primer (RACE 3) situated in the first exon sequenced (Table 1). A poly-cytosine tail was added to the transcribed cDNA using recombinant terminal transferase (Roche). Nested PCR was performed using a poly-guanosine forward primer and 2 gene-specific reverse primers (RACE 2 and RACE 1 respectively) as described in Table 1.
TgPREX polymerase domain cloning and expression
From the alignment of the predicted PREX proteins in different apicomplexan organisms (data not shown), the exonuclease-polymerase domain of the T. gondii PREX was identified and a 3060 bp region encompassing the entire domain was amplified from T. gondii tachyzoite cDNA using primers Tg_polymeraseF and R20.2 (Table 1). The fragment was cloned into the pGEM-T Easy vector and digested with NheI (shown in the Tg_polymeraseF primer in underlined italics in Table 1) and SacI (restriction enzyme site in the pGEM-T Easy plasmid) enzymes, and subsequently ligated into the pET28a+ vector. In the resulting construct, TgPREX is fused to an N-terminal hexa-histidine tag and expressed under the control of the T7 promoter.
The pET28a-TgPREX polymerase plasmid was used for transformation of the E. coli BL21-AI (Invitrogen) and cells were grown at 37°C in 3 L of LB medium containing 0·1% glucose and 50 μg/ml carbenicillin; L-arabinose (0·2% final concentration) was added at OD600=~0·6. After further incubation for 4 h at 37°C, cells were harvested by centrifugation and resuspended in 60 ml of buffer A [50 mm Tris-HCl (pH 8·0), 50 mm NaCl, 1 mm EDTA, 1 mm PMSF, 2 mm B-mercaptoethanol, 2 mm benzamidine and 10% (w/v) glycerol], and adjusted to 500 μg/ml lysozyme prior to storage at −80°C.
The cell suspension was thawed, immediately supplemented with 0·1× protease inhibitor cocktail (EMD Biosciences) and sonicated on ice. The following procedures were carried out at 4°C. After centrifugation at 20 000 g for 30 min, the supernatant was preserved, and the cell pellet resuspended in 15 ml of buffer A; the sonification and centrifugation steps were repeated and the supernatant fractions were combined (Fraction I). Fraction I was loaded onto a DEAE cellulose (Sigma) column (4·9 cm2×4 cm) equilibrated in buffer A and the column was washed with 100 ml of buffer A. Proteins were eluted by using a 120 ml linear gradient of 50–500 mm NaCl in buffer A. Fractions (5 ml) containing DNA polymerase activity were pooled (Fraction II) and dialysed against buffer B [50 mm Tris-HCl (pH 8·0), 50 mm NaCl, 1 mm EDTA, 0·1 mm PMSF, 2 mm β-mercaptoethanol, 2 mm benzamidine and 10% (w/v) glycerol]. Following dialysis, Fraction II was applied to a P-11 (GE Healthcare, Piscataway, NJ) column (1·8 cm2×7 cm) equilibrated in buffer B. The column was washed with 60 ml of buffer B, followed by elution with a 60 ml linear gradient of 50–500 mm NaCl in buffer B. The fractions (2·5 ml) were analysed in 10% SDS-polyacrylamide gels, and fractions containing DNA polymerase activity were pooled (Fraction III) and dialysed against buffer C [50 mm sodium phosphate (pH 8·0), 250 mm NaCl, 10 mm imidazole, 1 mm β-mercatoethanol, 2 mm benzamidine and 5% (w/v) glycerol]. Dialysed Fraction III was loaded onto a Qiagen nickel-nitrilotriacetic acid (Ni-NTA) affinity column (1·77 cm 2×3 cm) equilibrated in buffer C. The column was washed with 25 ml of buffer C and then with a 30 ml linear gradient of 10–300 mm imidazole (pH 8·0) in buffer C. Fractions (2·5 ml) containing TgPREX polymerase proteins, identified by electrophoresis in 10% SDS-polyacrylamide gels, were pooled (fraction IV). After dialysis against storage buffer [50 mm Tris-HCl (pH 7·4), 1 mm DTT, 30 mm NaCl and 10% (w/v) glycerol], the preparation was concentrated using an Amicon filter unit (MW cut-off 50 000). Aliquots were made and stored at −80°C. The purified TgPREX polymerase protein (Fraction IV) appeared homogenous, as assessed in a 10% SDS-polyacrylamide gel followed by the Commassie Blue G-250 staining. Protein concentration was determined by the Bradford reaction (Bradford, Reference Bradford1976) using the Bio-Rad Protein Assay with bovine serum albumin as a standard.
Mass spectrometric analysis of the expressed protein
The purified protein was used for trypsin digestion (Bridges et al. Reference Bridges, Pitt, Hanrahan, Brennan, Voorheis, Herzyk, de Koning and Burchmore2008). Tryptic peptide samples were separated on an LC system (Famos/Switchos/Ultimate, LC Packings) before being analysed by electrospray ionisation (ESI) Mass Spectrometry (MS) on a Q-STAR® Pulsar i hybrid LC/MS/MS System. Peptide separation was performed on a Pepmap C18 reversed phase column (LC Packings), using a 5–85% v/v acetonitrile gradient (in 0·5% v/v formic acid) run over 45 min. The flow rate was maintained at 0·2 μl/min. Mass spectrometric analysis was performed using a 3-sec survey MS scan followed by up to 4 MS/MS analyses of the most abundant peptides (3 sec per peak) in Information Dependent Acquisition (IDA) mode, choosing 2+ to 4+ ions above threshold of 30 counts, with dynamic exclusion for 120 sec.
Data generated from the Q-STAR® Pulsar i hybrid mass spectrometer was analysed using Applied Biosystems Analyst QS (v1.1) software and the automated Matrix Science Mascot Daemon server (v2.1.06). Protein identifications were assigned using the Mascot search engine, which gives each protein a probability-based MOWSE score. In all cases variable methionine oxidation was allowed in searches. An MS tolerance of 1·2 Da for MS and 0·4 Da for MS/MS analysis was used.
DNA polymerase activity assay
DNA polymerase activity was assayed as described (Glick et al. Reference Glick, Anderson and Loeb2002). Briefly, the activity was measured at 37°C for 15 min in 10 μl reaction mixtures containing 1 μg of activated calf thymus DNA, 25 μm each dNTP, 1 μCi α[32P] dTTP (3000 Ci/mmol) (PerkinElmer, Waltham, MA, USA), and 1 μl of enzyme in 30 mm Tris-HCl, pH 7·0, 10 mm MgCl2, 1 mm DTT, 250 μg BSA, and 2·5% glycerol. The reaction was terminated by the addition of 100 μl of 0·1 m sodium pyrophosphate/0·05 m EDTA. An aliquot (100 μl) of the reaction mixture was transferred to a well in a 96-microwell® plate (Biodyne® B; NUNC™) mounted on a 96-vacuum manifold (Beckman Coulter, Fullerton, CA, USA). The plates were washed 3 times with 250 μl of 0·1 m sodium pyrophosphate. The filter was removed from the plate, dried, and the amount of radioactivity associated with the filter was quantified by phosphorimager analysis using ImageQuant® software (GE Healthcare, Piscataway, NJ).
Software
Vector NTI Advance™ suite 9 and 10 (Infomax 2003 from Invitrogen) was used for sequence analysis. The exon-intron structure of the Tgprex gene was created using GSDS, a gene structure display software (http://gsds.cbi.pku.edu.cn/) (Guo et al. Reference Guo, Zhu, Chen and Luo2007). SignalP 3.0 Server (http://www.cbs.dtu.dk/services/SignalP/) (Bendtsen et al. Reference Bendtsen, Nielsen, von Heijne and Brunak2004; Nielsen et al. Reference Nielsen, Engelbrecht, Brunak and von Heijne1997; Nielsen and Krogh, Reference Nielsen and Krogh1998), SIG-Pred (http://www.bioinformatics.leeds.ac.uk/prot_analysis/Signal.html), SIGFIND (http://139.91.72.10/sigfind/sigfind.html), PrediSi (Hiller et al. Reference Hiller, Grote, Scheer, Munch and Jahn2004) and ChloroP 1.1 Server (http://www.cbs.dtu.dk/services/ChloroP/#submission) (Emanuelsson et al. Reference Emanuelsson, Nielsen and von Heijne1999) were used for signal and transit peptide identification of TgPREX protein.
RESULTS
Identification of a PREX homologue in ToxoDB
Two genes (55.m04962/TGME49_061920 and 55.m04960/TGME49_061800) were identified in ToxoDB (release 4 and 5 respectively) on chromosome VIIb of T. gondii genomic sequence on contigs 994 270 and 994 314 in the antisense strand (Kissinger et al. Reference Kissinger, Gajria, Li, Paulsen and Roos2003). The coding sequence of this region (1 488 133 to 1 515 551 bp) on chromosome VIIb was clarified by PCR amplification from T. gondii cDNA using primer pairs delineated in Fig. 1A.
Following sequencing, it appeared that a single gene homologous to Pfprex is present in T. gondii cDNA; thereafter, we call this gene Tgprex. The Tgprex gene comprises of 20 exons as shown in Fig. 1A. The 5′ end of the ORF was elucidated by 5′ RACE (Fig. 1B). The initiation codon of the ORF (Fig. 1C) was confirmed by comparing the 2345 bp long sequenced PCR product from the cDNA with that of genomic DNA (Fig. 1C). Sequencing of the 1766 bp long cDNA PCR product in comparison to genomic DNA PCR (Fig. 1D) confirmed the connection between the putative primase-helicase and the polymerase region of the Tgprex gene. The putative domain structure of the translated TgPREX protein is represented in Fig. 1E.
In summary, a 7740 bp long ORF, (TgondiiORF7740/Tgprex, GenBank Accession no. FJ665392) was identified from the sequenced region of chromosome VIIb (between 1,491,400 and 1,512,118). A sequencing gap between contigs 994 270 and 994 314 in the database was sequenced from both the genomic DNA and the cDNA of the RH strain of the organism and a 146 bp long sequence was subsequently deposited in the database.
Prediction of localization of TgPREX in silico
The TgPREX translated protein sequence possessed a primase, helicase and polymerase homologous domain structure (Fig. 1E) similar to that of the PREX protein of Plasmodium. The peptide region of the TgPREX protein that separated the primase-helicase and the polymerase domains has no similarity to the spacer region occupying the same region of the PfPREX predicted peptide sequence.
T. gondii uses the same general mechanism to mediate transport of proteins into the apicoplast as Plasmodium (He et al. Reference He, Striepen, Pletcher, Murray and Roos2001). A bipartite leader peptide, including a primary secretory domain followed by a secondary plastid transit domain, can be predicted by specialized tools including PlasmoAP (Foth et al. Reference Foth, Ralph, Tonkin, Struck, Fraunholz, Roos, Cowman and McFadden2003) and PATS (Waller et al. Reference Waller, Keeling, Donald, Striepen, Handman, Lang-Unnasch, Cowman, Besra, Roos and McFadden1998; Zuegge et al. Reference Zuegge, Ralph, Schmuker, McFadden and Schneider2001) in PlasmoDB. These programmes, however, failed in systemically identifying the apicoplast targeted proteins in T. gondii (Harb et al. Reference Harb, Chatterjee, Fraunholz, Crawford, Nishi and Roos2004). For example, known T. gondii nuclear encoded apicoplast proteins (e.g. acyl carrier protein [AAC63956], beta-hydroxyacyl-ACP dehydratase [AAC72191], small ribosomal protein S9 [AAC63957], large ribosomal protein L28 [AAC63958] (Waller et al. Reference Waller, Keeling, Donald, Striepen, Handman, Lang-Unnasch, Cowman, Besra, Roos and McFadden1998) and a putative ferredoxin NADP+ oxidoreductase [CAC15394] (Vollmer et al. Reference Vollmer, Thomsen, Wiek and Seeber2001) were all unrecognized. For each of these proteins, however, the signal peptide component was recognized by SignalP 3.0 and the plastid targeting peptide following the signal peptide was recognized by ChloroP 1.1.
The N-terminal end of the TgPREX protein was analysed sequentially for the presence of a bipartite leader peptide. A 70 amino acid long N-terminal sequence of TgPREX was used as a query in PrediSi and SIGFIND software. Amino acid residues from 30 to 70 were used as a query in SignalP 3.0 as the maximum length of sequence input is restricted to 30 in this programme. All the analyses predicted TgPREX as an apicoplast-localized protein similar to its homologous counterpart PfPREX whose N-terminus was shown to direct GFP to the apicoplast (Seow et al. Reference Seow, Sato, Janssen, Riehle, Mukhopadhyay, Phillips, Wilson and Barrett2005). TgPREX appears to possess a putative, comparatively long N-terminal signal peptide, 60 to 61 amino acids in length. The cleavage sequence has been identified within the multiple serine residues (VLS-SS or VLSS-SS). The signal peptide was followed by a 61-amino acid long, putative plastid transit peptide as identified by the ChloroP1.1 programme.
Analysis of the PREX polymerase
The recombinant polymerase domain of the TgPREX, with a predicted mass of 114 kDa was expressed in E. coli. The histidine-tagged protein was purified by Ni2+ affinity chromatography. The protein was analysed by SDS-PAGE and the identity was confimed by mass spectrometry. The identified peptides are detailed in the Supplementary Data 1 (Online version only).
The purified TgPREX polymerase recombinant protein (Fraction IV) (Fig. 2A) was tested for its DNA polymerase activity. Activity was optimal at a temperature of 50°C (Fig. 1C) and at pH 7·0 (Fig. 1D). The enzyme required Mg2+ ions for activity (Fig. 1B), which reached the optimum at 5 mm MgCl2 (Fig. 1E).
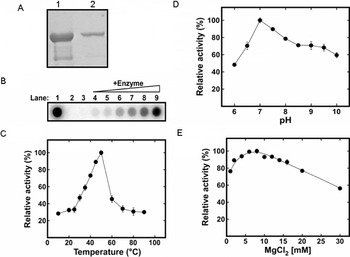
Fig. 2. SDS-PAGE and functional analysis of the purified TgPREX polymerase protein. The SDS-PAGE of purified TgPREX revealed a 114 kDa protein band in purified (2) followed by concentrated (1) protein samples. Functional properties of the purified TgPREX protein. (B) For TgPREX DNA polymerase activity Mg2+ ions are required. In the absence of Mg2+ ions, the amount of incorporated [α]32P-dTTP by the TgPREX to the activated calf thymus DNA is similar to that without the enzyme addition (Lanes 3 and 2, respectively). The amounts of incorporated [α]32P-dTTP to the activated calf thymus DNA increases as the concentrations (3·1, 6·3, 12·5, 25, 50 and 100 nm, respectively) of TgPREX increases in the reaction (Lanes 4–9, respectively). The DNA polymerase activity of E. coli Klenow (1 unit) is also shown as a control (Lane 1). (C) TgPREX polymerase activity measured as a function of MgCl2 concentration; (D) TgPREX polymerase activity measured as a function of pH; (E) TgPREX polymerase activity measured as a function of temperature. Error bars represent the standard deviation of 3 independent experimental determinations.
DISCUSSION
PREX, the plastid replication and repair enzyme complex of apicomplexan parasites was first identified in Plasmodium. Homology to DNA primase, helicase and polymerase in other systems indicated that the gene codes for all of these key functions. The presence of a predicted plastid targeting sequence and the fact that a similar sequence in P. falciparum guides marker proteins to the apicoplast suggest that PREX is instrumental in the replication of apicoplast DNA.
Among the annotated genes related to Tgprex in ToxoDB, TGME49_061800 expression is linked to one EST (Expressed Sequence Tag) and 7.6 normalized SAGE (serial analysis of gene expression) tag counts (Gajria et al. Reference Gajria, Bahl, Brestelli, Dommer, Fischer, Gao, Heiges, Iodice, Kissinger, Mackey, Pinney, Roos, Stoeckert, Wang and Brunk2008). There are no such data related to the TGME_061920 gene. Mass spectrometry analysis has identified 2 peptide sequences (ARPLSPEHSALNESAGCAR and LFLESATPVPHAQILTFR) (Xia et al. Reference Xia, Sanderson, Jones, Prieto, Yates, Bromley, Tomley, Lal, Sinden, Brunk, Roos and Wastling2008) which correspond to peptides in exon 4 and 5 of the sequenced Tgprex gene respectively. The lack of good EST coverage for this gene probably corresponds to a low level expression as documented in ToxoDB at the 32·2 and 25·2 percentile for TGME49_061800 and TGME_061920, respectively. A similarly low level of expression was also observed in P. falciparum (Le Roch et al. Reference Le Roch, Zhou, Blair, Grainger, Moch, Haynes, De, Holder, Batalov, Carucci and Winzeler2003).
BLAST searches identified full length PREX orthologues in other Plasmodium species, in Theileria parva [XP_765913] and Theileria annulata [XP_954352] and in Babesia bovis (XP_001610510) (Table 2) (Alignment in Supplementary Data 2 – Online version only). Homologues representing fragments of the total predicted protein were also identified in Babesia bigemina, Eimeria tenella, Neospora caninum and Sarcocystis neurona where respective genome sequencing is incomplete. Interestingly, no homologous protein was found in the Cryptosporidium species. Genome sequencing also suggests that Cryptosporidium hominis (Xu et al. Reference Xu, Widmer, Wang, Ozaki, Alves, Serrano, Puiu, Manque, Akiyoshi, Mackey, Pearson, Dear, Bankier, Peterson, Abrahamsen, Kapur, Tzipori and Buck2004) and Cryptosporidium parvum (Abrahamsen et al. Reference Abrahamsen, Templeton, Enomoto, Abrahante, Zhu, Lancto, Deng, Liu, Widmer, Tzipori, Buck, Xu, Bankier, Dear, Konfortov, Spriggs, Iyer, Anantharaman, Aravind and Kapur2004) lack an apicoplast.
Table 2. PREX homologous genes in other apicomplexa

Interestingly, the prex gene of T. gondii differs from that of P. falciparum in that it is interrupted by 20 introns. The intron footprint of eukaryotic, nuclear-encoded genes with possible prokaryotic cyanobacterial ancestry has made a significant contribution to our understanding of gene evolution. The presence of introns in such genes suggests the insertion of introns into pre-assembled genes of eukaryotes late in evolution (‘intron late’) as the ancestral cyanobacterial genes were devoid of introns. Similar comparison of ribosomal protein S9, L28 and acyl carrier protein genes between P. falciparum and T. gondii suggests a process of continuous intron insertion during evolution even after the divergence of Plasmodium and T. gondii from their common ancestor (Schaap et al. Reference Schaap, van Poppel and Vermeulen2001).
The polymerase function of this putative protein, identified only in apicoplast-bearing apicomplexans, was confirmed in T. gondii. The nuclear encoded PREX appears to serve as a replicative enzyme for the 35 kb genome of the apicoplast. The participation of nuclear-encoded proteins in apicoplast replication has also been suggested for DNA gyrase subunits A and B (Dar et al. Reference Dar, Sharma, Mondal and Dhar2007; Raghu Ram et al. Reference Raghu Ram, Kumar, Biswas, Kumar, Chaubey, Siddiqi and Habib2007), a DNA ligase and 2 hypothetical proteins bearing similarities with DNA-repair proteins (Dahl and Rosenthal, Reference Dahl and Rosenthal2008).
Given that inhibitors of other enzymes involved in plastid replication (e.g. inhibitors of DNA gyrase) are toxic to apicomplexan parasites the PREX protein complex may be considered a target for chemotherapy. This may apply particularly to the primase and polymerase domains since the helicase domain has substantial homology to the Twinkle helicase that is active in mitochondria of most eukaryotic species.
We thank Dr Richard J. S. Burchmore of Sir Henry Wellcome Functional Genomics Facility, Institute of Biomedical and Life Sciences, University of Glasgow, for performing the MS analysis of protein in our study. A.M. was funded by the Wellcome Trust as part of their “Molecular Functions in Disease” programme.






