Introduction
Rice, a staple food crop, is conventionally cultivated under flooded anaerobic conditions (Castaneda et al., Reference Castaneda, Bouman, Peng and Visperas2002; Chan et al., Reference Chan, Zainudin, Saad and Azmi2012). Efforts are being made to grow rice under semi-irrigated (aerobic) conditions (Dharmappa et al., Reference Dharmappa, Doddaraju and Malagondanahalli2019). Irrigation practices like furrow and multiple inlet irrigation, alternate wetting and drying and overhead sprinklers have gained interest to optimize water use (Bouman et al., Reference Bouman, Lampayan and Tuong2007; Girsang et al., Reference Girsang, Quilty, Correa, Sanchez and Buresh2019; Graham-Acquaah et al., Reference Graham-Acquaah, Siebenmorgen, Reba, Massey, Mauromoustakos, Adviento-Borbe, January, Burgos and Baltz-Gray2019).
Semi-irrigated rice experiences desiccation stress between irrigations which affects its growth and productivity (Borrell et al., Reference Borrell, Garside and Fukai1997; Dharmappa et al., Reference Dharmappa, Doddaraju and Malagondanahalli2019). Thorough knowledge of stress tolerance mechanism can help in crop improvement and management. Divergent genes and pathways associated with desiccation stress are reported (Li et al., Reference Li, Nong, Solanki, Liang, Xie, Liu, Li, Wang, Yang and Li2016; Parvathi and Nataraja, Reference Parvathi, Nataraja and Senthil-Kumar2017; Priya et al., Reference Priya, Dhanker, Siddique, Hanumantha Rao, Nair, Pandey, Singh, Varshney, Prasad and Nayyar2019; Sharma et al., Reference Sharma, Jain and Khurana2019; Azzouz-Olden et al., Reference Azzouz-Olden, Hunt and Dinkins2020). In transgenic plants, stress-inducible genes such as LEA, HSPs, AREB/ABFs, bZIPs contributed to enhanced stress tolerance (Ali et al., Reference Ali, Bano and Fazal2017). In model plants, epigenetic mechanisms such as DNA methylation, and histone modifications, involved in stress responses are reported (Munshi et al., Reference Munshi, Ahuja, Bahadur, Bahadur, Venkat Rajam, Sahijram and Krishnamurthy2015; Mozgova et al., Reference Mozgova, Mikulski, Pecinka, Farrona, Alvarez-Venegas, De-la-Peña and Casas-Mollano2019; Godwin and Farrona, Reference Godwin and Farrona2020); however, studies in field crops are limited (Wang et al., Reference Wang, Qin, Sun, Wang, Xu, Li and Fu2016).
DNA methylation is one of the important epigenetic mechanisms in mammals and plants (Meyer, Reference Meyer2015; Schübeler, Reference Schübeler2015). In higher eukaryotes, DNA methylation determines chromatin structure, regulates replication and gene expression (Tirado-Magallanes et al., Reference Tirado-Magallanes, Rebbani, Lim, Pradhan and Benoukraf2017; Varotto et al., Reference Varotto, Tani, Abraham, Krugman, Kapazoglou, Melzer, Radanović and Miladinović2020). DNA methylation is generally associated with suppression of gene expression while demethylation results in gene activation (Zemach et al., Reference Zemach, Kim, Silva, Rodrigues, Dotson, Brooks and Zilberman2010). In plants, DNA methylation regulates gene expression under stress (Garg et al., Reference Garg, Chevala, Shankar and Jain2015). The CpG dinucleotide is the predominant site for DNA methylation in vertebrates (Bird, Reference Bird1987), and CpG clusters are referred to as CpG islands (CGI) (Deaton and Bird, Reference Deaton and Bird2011). In plants, promoter methylation suppresses gene expression in a tissue-specific manner (Finnegan et al., Reference Finnegan, Genger, Peacock and Dennis1998; Zilberman et al., Reference Zilberman, Gehring, Tran, Ballinger and Henikoff2007). A correlation between promoter CGI and transcription initiation in vertebrates exists (Deaton and Bird, Reference Deaton and Bird2011). Our study, using 5′-Azacitidine (methylation inhibitor) and rice genotypes (AC39020 and BPT5204), contrasting for drought tolerance indicated that CGI could be associated with the methylation pattern in the promoter region, and methylation might modulate gene expression under salinity stress (Sapna and Nataraja, Reference Sapna and Nataraja2016).
In rice under drought, several genes corresponding to differentially methylated regions are altered (Wang et al., Reference Wang, Qin, Sun, Wang, Xu, Li and Fu2016). Drought induces alterations in DNA methylation locus and differential patterns in plants with variety, tissue and stress specificity (Wang et al., Reference Wang, Pan, Zhao, Dwivedi, Zhu, Ali, Fu and Li2011; Kinoshita and Seki, Reference Kinoshita and Seki2014). In Arabidopsis, drought during vegetative stage induces phenotypic and DNA methylation plasticity (Van-Dooren et al., Reference Van-Dooren, Silveira, Gilbault, Jiménez-Gómez, Martin, Bach, Tisné, Quadrana, Loudet and Colot2020). Stress-responsive transcription factors of Zinc-finger family showed a correlation between methylation and gene expression pattern under stress (Ahmad et al., Reference Ahmad, Farman, Waseem, Rana, Nawaz, Rehman, Abbas, Baloch, Akrem, Huang and Zhang2019).
In this study, we examined DNA methylation pattern using methylation-sensitive randomly amplified polymorphic DNA (MS-RAPD) approach, and expression of four genes in rice genotype, IR64 (Lenka et al., Reference Lenka, Katiyar, Chinnusamy and Bansal2011) grown under different soil water status. We observed drought-induced methylation, which might be one of the major mechanisms associated with drought responses in rice.
Materials and methods
Plant material and stress treatment
Two days old rice seedlings (Oryza sativa subsp. indica), variety, IR64, a lowland cultivar developed by the International Rice Research Institute in the Philippines, were raised in polybags in a containment facility under controlled conditions for 15 d. Healthy seedlings were then transplanted to pots (approximately 30 kg capacity) containing garden soil and maintained under greenhouse conditions. One set of plants were grown under puddled condition while other sets were maintained at 100% soil field capacity (FC) by gravimetric approaches (Karaba et al., Reference Karaba, Dixit, Greco, Aharoni, Trijatmiko, Marsch-Martinez and Pereira2007). A subset of plants maintained at 100% FC was exposed to drought stress (60% FC), 30 d post transplanting (at vegetative stage) using the gravimetric approach. The three sets of plants (puddled, 100 and 60% FC) were grown under greenhouse conditions. Physiological and biochemical parameters were recorded to assess the stress effect, and the fourth leaf of every tiller was collected 14 d post stress imposition for further studies.
Estimation of plant water status
The degree of stress was assessed using relative water content (RWC) which was measured in 50 d old plants using a standard protocol (Barrs and Weatherley, Reference Barrs and Weatherley1962). Leaf discs collected were floated in water for 5 h at 28°C after recording the fresh weight and then the turgid weight was determined. The dry weight was recorded after drying the samples for 72 h at 80°C in a hot air oven.
Estimation of total chlorophyll
About 100 mg leaf tissue was soaked in 10 ml mixture of 80% (v/v) acetone and dimethyl sulfoxide (1:1) to extract chlorophyll. Total chlorophyll was estimated according to Hiscox and Israelstam, (Reference Hiscox and Israelstam1979). The absorbance of the extract was measured at 652 nm using UV visible spectrophotometer (UV-VIS 2450, Shimadzu Corporation, Kyoto, Japan). Percentage reduction in chlorophyll content over puddled conditions was calculated.
Measurement of gas exchange parameters
Gas exchange parameters were recorded by a portable photosynthesis system, infrared gas analyser-IRGA (LICOR Inc., Lincoln, Nebraska, USA) (Nataraja and Jacob, Reference Nataraja and Jacob1999). Three readings were taken in the same leaf (4th leaf). All the gas exchange assessment was made at a light intensity of 1200 μmol/m2/s, ambient CO2 concentration of 420 μmol/mol and 32°C temperature. The relative air humidity in the leaf chamber was maintained at 70–80%.
Genomic DNA extraction and MS-RAPD
DNA was extracted by the cetyltrimethylammonium bromide method as described by Doyle and Doyle (Reference Doyle and Doyle1987). MS-RAPD analysis was performed by overnight digestion of 2 μg genomic DNA with methylation-insensitive restriction enzyme MspI (SibEnzyme) and methylation-sensitive restriction enzyme HpaII (SibEnzyme). The digested DNA was further subjected to PCR using random primers. Out of 40 primers, 10 primers that gave reproducible results for two replications were selected for further analysis. The sequences (5′→3′) of the primers utilized were GGAAGCCAAC (P15), CACAGCTGCC (R2), GTCTACGGCA (R6), GGACAACCAG (R15), ACGGCAAGGA (R20), GGCAGGCTGT (T7), GGAGCCTCAG (X11), GGGCCAATGT (Y16), GACGTGGTGA (Y17), and GTGGAGTCAG (Y18).
The RAPD-PCR was performed in a 10 μl reaction mixture containing 100 ng of DNA template, 1X buffer, 0.2 mM dNTP each, 1.5 mM MgCl2 and 1U Taq DNA Polymerase (Invitrogen by Life Technologies, California, USA). The amplifications were performed in a thermal cycler (ProFlex PCR systems by Applied Biosystems, California, USA) programmed as follows: 3 min at 95°C, followed by 45 cycles of 95°C (1 min), 35°C (1 min) and 72°C (90 s), and a final extension of 10 min at 72°C. The PCR products were resolved on 1.5% (w/v) agarose gel and the amplified product was detected by ethidium bromide staining. Based on the procedure described by Erturk et al. (Reference Erturk, Agar, Arslan, Nardemir and Sahin2014), polymorphism in the MS-RAPD profile was identified as the disappearance of a normal band and appearance of a new band in the treated sample compared to control.
CpG island detection
The gene IDs were obtained from the Rice Annotation Project database (RAP-DB). The promoter sequences (2 kb) were obtained from the plant promoter database (http://ppdb.agr.gifu-u.ac.jp/ppdb/cgi-bin/index.cgi). These sequences were submitted to the tool EMBOSS (https://www.ebi.ac.uk/Tools/seqstats/emboss_newcpgreport) and CpG islands from 2 kb upstream region of the gene were extracted.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from leaf tissues by a modified phenol chloroform protocol (Sajeevan et al., Reference Sajeevan, Shivanna and Nataraja2014). About 2 μg RNA was converted to cDNA (Invitrogen Super ScriptTM First-Strand Synthesis, California, USA). The qRT-PCR analysis was performed using a few drought-responsive genes (DRGs) with or without CpG islands, identified using in silico analysis. qRT-PCR was carried out using a SYBR green supermix (iTaq™ Universal SYBR® Green Supermix, BIO-RAD, California, USA) in a CFX 96 real-time touch system (BIO-RAD). The reaction was performed in a 20 μl volume and the conditions were as follows: 95°C for 3 min, followed by 35 cycles (95°C for 10 s, 60°C for 15 s, 72°C for 30 s). A no template control was included in each reaction. Primers used in the expression studies are presented in Table 2. The qRT-PCR results were normalized to Ubiquitin from the respective sample and fold change calculated by using 2(−ΔΔC(T)) method (Livak and Schmittgen, Reference Livak and Schmittgen2001). The transcript levels in 100 and 60% FC plants were expressed in relation to puddled plants.
Statistical analysis
The results obtained from the physiological experiments and qRT-PCR were represented as mean ± standard error (SEM). The effects of drought on the physiological parameters were determined using one-factor ANOVA at P < 0.05. The significant difference for the expression data was tested using Student's t-test with minimal significance at 0.05 level.
Results
Assessing the effect of drought stress
The rice plants exposed to drought stress showed leaf rolling and wilting phenotype 14 d post stress (Fig. 1). The stress effect on the plants subjected to different regimes of moisture stress was evaluated by analysing RWC, photosynthetic rate, stomatal conductance and chlorophyll content. There was a significant reduction (P < 0.05) in RWC in plants maintained at 100% (77.2%) and 60% (42.4%) FC, compared to puddled (89%) plants (Fig. 2(a)). The photosynthetic rates were significantly reduced (P < 0.05) from 14 μmol/m2/s in puddled to 12 and 10 μmol/m2/s under 100 and 60% FC, respectively (Fig. 2(b)). The stomatal conductance also showed significant reductions in 100% FC (0.35 mol/m2/s) and 60% FC (0.31 mol/m2/s) compared to puddled (0.43 mol/m2/s) (Fig. 2(c)) plants. As expected, there was a reduction in chlorophyll content under stress. At 100 and 60% FC, a significant reduction (P < 0.05) of 20 and 51%, respectively, was noticed in the chlorophyll content (Fig. 2(d)). These physiological parameters indicated that the rice plants grown under 100 and 60% FC were experiencing desiccation stress.

Fig. 1. Phenotype of rice genotype IR64 under different soil water status. Photographs were taken 14 d after drought stress imposition.

Fig. 2. Evaluation of drought stress by assessment of (a) relative water content, (b) photosynthetic rate, and (c) stomatal conductance, (d) per cent reduction in total chlorophyll content of rice plants grown under different levels of soil water status. The significance at levels P < 0.05 is indicated as alphabets.
DNA methylation patterns
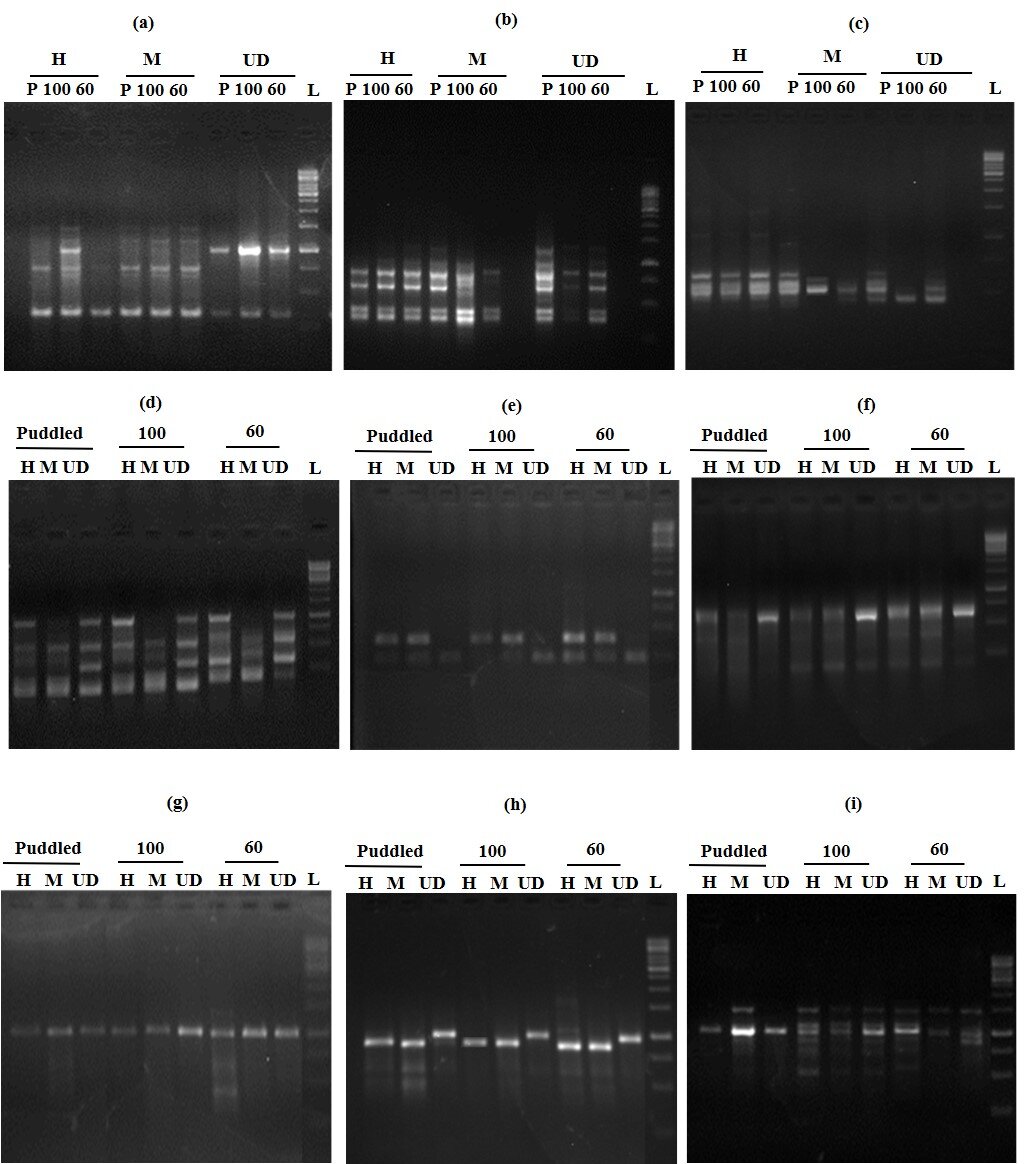
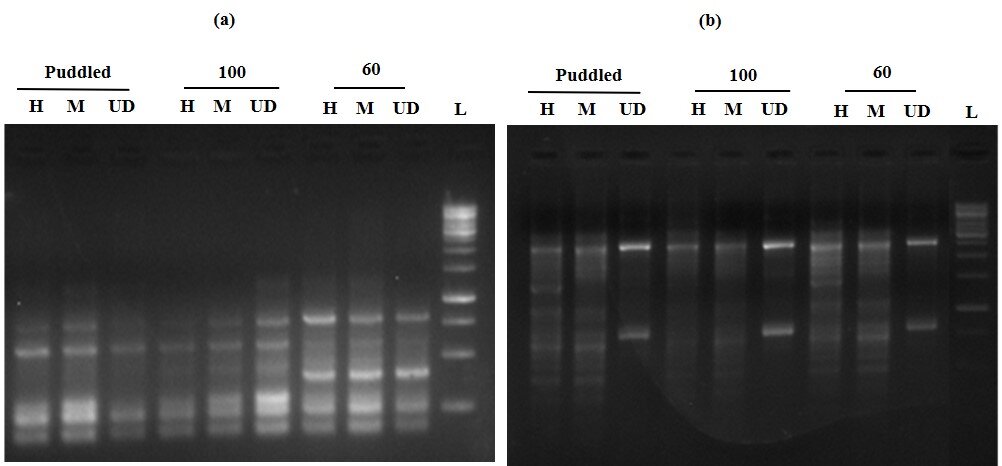
MS-RAPD PCR with primer Y18 revealed a similar number of fragments in DNA digested with HpaII and MspI under puddled and 60% soil FC, with an increase in the number of fragments in 60% FC compared to puddled plants (Fig. 3). However, DNA samples from plants subjected to 100% FC, digested with HpaII showed a reduced number of fragments compared to MspI digested DNA. This suggests that the plants subjected to 60% FC have increased methylation. Most of the primers used for this analysis showed a similar banding pattern under severe stress. The polymorphic bands observed under moisture stress with each primer are represented in Table 1. The increase in the number of bands in HpaII digested DNA suggests that there was an increase in methylation at 60% FC, whereas the reduced number of bands in 100% FC and puddled indicated less methylation.

Fig. 3. MS-RAPD using RAPD primer, Y18 in the leaf tissue of rice plants grown under different water status. The samples were digested with HpaII and MspI and subjected to RAPD-PCR. ‘L’ is for ladder. The variation in methylation pattern as described in the text is marked with by asterisks.
Table 1. MS-RAPD profile showing the total number of polymorphic bands in rice plants grown under different levels of soil water status

100 and 60 refer to 100 and 60% FC, respectively; H and M refer to digestion with HpaII and MspI, respectively.
Gene expression
Based on the in-silico analysis using the existing microarray datasets on rice genotypes subjected to drought stress, a few differentially regulated genes having a cut-off value of 1.5 were identified. The expression levels of four different genes with or without CpG islands (Table 2) were analysed using qRT-PCR (Fig. 4). Analysis of the promoter region of protein phosphatase 2C (PP2C) gene indicated the presence of one CpG island. The expression of PP2C was low (<1.5-fold) under 100 and 60% FC. The phenylalanine ammonia-lyase (PAL) with three CpG islands showed lower expression in 60% FC (<1-fold) and 100% FC (<1-fold). Heat Shock protein 70 (HSP70) and DEAD-box ATP-dependent RNA helicase 25 (RH25) do not possess any CpG island in their promoter region. The expression of HSP70 under 60% FC (>2-fold) was higher than plants maintained at 100% FC (<1-fold). The RH25 gene showed a significant increase in the expression under 100% (0.45-fold) and 60% (6.91-fold) FC. The study provided a lead but not conclusive evidence, on the relevance of CpG islands and its association with gene expression in rice.

Fig. 4. qRT-PCR expression analysis of four different genes in the leaf tissue of rice plants grown under different soil water status. Ubiquitin was used as the internal control and the expression in plants subjected to 100 and 60% FC was indicated as fold change over control (puddled). ‘*’ indicates significance at P < 0.05.
Table 2. Primers used for gene expression studies in rice plants

Discussion
DNA methylation is one of the epigenetic modifications that take place in the nucleosome at different levels through reversible biochemical reactions (Mozgova et al., Reference Mozgova, Mikulski, Pecinka, Farrona, Alvarez-Venegas, De-la-Peña and Casas-Mollano2019). It plays a key role in regulating gene expression required for modulating plant growth and development, and response to different environmental factors. We used rice, one of the most drought-sensitive and an important food crop traditionally grown under well-irrigated conditions (Wang et al., Reference Wang, Qin, Sun, Wang, Xu, Li and Fu2016), for DNA methylation analysis. There are reports on DNA methylation and epigenetic mechanism in indica rice under drought stress to understand the complex regulatory mechanisms involved in drought responses (Garg et al., Reference Garg, Chevala, Shankar and Jain2015; Wang et al., Reference Wang, Qin, Sun, Wang, Xu, Li and Fu2016). We used MS-RAPD approach to examine DNA methylation pattern in the leaf tissue of rice genotype, IR64 grown under different soil moisture status. As reported by Erturk et al. (Reference Erturk, Agar, Arslan, Nardemir and Sahin2014), we used methylation-sensitive (HpaII) and insensitive (MspI) restriction enzymes for the methylation analysis. This technique has been successfully used in crop plants such as cotton and maize to study DNA methylation (Erturk et al., Reference Erturk, Agar, Arslan, Nardemir and Sahin2014; Karaca et al., Reference Karaca, Aydin and Ince2019). A similar approach was also used to understand the epigenetic effects of salinity on barley (Demirkiran et al., Reference Demirkiran, Marakli, Temel and Gozukirmizi2013). Our analysis indicated increased methylation under severe drought stress (60% FC) compared to the plants grown under puddled conditions, and the data presented here are similar to the previous reports which state that abiotic stresses tend to modify the methylation status of genomic DNA (Correia et al., Reference Correia, Valledor, Hancock, Jesus, Amaral, Meijón and Pinto2016; Wang et al., Reference Wang, Qin, Sun, Wang, Xu, Li and Fu2016).
Global methylation or demethylation may be correlated to gene expression under abiotic stresses (Wang et al., Reference Wang, Qin, Sun, Wang, Xu, Li and Fu2016). Zemach et al. (Reference Zemach, Kim, Silva, Rodrigues, Dotson, Brooks and Zilberman2010) suggested that the genes which are least expressed are most likely to be methylated and highly expressed genes are less methylated. Whole-genome methylome analysis revealed that the methylation, especially at the CpG islands in the promoter region, appeared to be related to tissue-specific expression in plants (Surdonja et al., Reference Surdonja, Eggert, Hajirezaei, Harshavardhan, Seiler, Von Wirén, Sreenivasulu and Kuhlmann2017). Surdonja et al. (Reference Surdonja, Eggert, Hajirezaei, Harshavardhan, Seiler, Von Wirén, Sreenivasulu and Kuhlmann2017) showed increased DNA methylation in the promoter region of HvCKX2.1 under severe drought stress. Drought induces cultivar-specific transgenerational and site-specific variations in DNA methylation in two contrasting rice genotypes, and the differential methylation was higher in the promoter regions of the genes responsive to different abiotic stress (Wang et al., Reference Wang, Pan, Zhao, Dwivedi, Zhu, Ali, Fu and Li2011; Zheng et al., Reference Zheng, Chen, Li, Lou, Xia, Wang, Li, Liu and Luo2013). Our previous study indicated that CpG islands might be associated with methylation under drought stress (Sapna and Nataraja, Reference Sapna and Nataraja2016). Promoter CpG methylation of genes may negatively regulate the binding of transcriptional factors or may bind methyl-CpG-binding domain proteins to regulate the gene expression (Su et al., Reference Su, Xia and Zhao2011). It is likely that methylation events affect the expression pattern of such genes associated with drought response.
We analysed the expression of four genes in leaf tissues collected from the plant grown under puddled, 100 and 60% FC. Since CpG island is the predicted target for methylation, we selected the genes with or without CpG islands in the promoter region. These genes included the enzymes involved in ABA signalling, protein phosphatase 2C(PP2C), PAL involved in the biosynthesis of phenolic compounds (Boudet, Reference Boudet2007; Yang et al., Reference Yang, Liu, Niu, Wang, Wan, Yang, Li, Wang and Wang2018), a chaperone (HSP70) and a helicase involved in RNA metabolism (RH25). At 60% FC, PP2C and PAL which have CpG islands in their promoter region showed lower levels of expression than HSP70 and RH25 which do not contain CpG island. A recent study on pineapple suggested that, DNA methylation at CpG islands in the promoter regions of a gene promoting somatic embryogenesis, SERK1 represses its expression (Luan et al., Reference Luan, Chen, Xie, He and He2020). Another study on transgenic flax crop showed that the gene chalcone synthase was more accessible to DNA methylation when located within CpG islands and the expression of the gene depended on the methylation status in these regions (Dzialo et al., Reference Dzialo, Szopa, Czuj and Zuk2017). The promoter of succinate dehydrogenase gene (sdh2-1) showed increased methylation but highly reduced gene expression on the eighth day of germination in the scutellum of maize seeds (Eprintsev et al., Reference Eprintsev, Fedorin, Karabutova and Igamberdiev2016). These reports as well as the present study provided a lead to argue that CpG islands may play a role in modulating the gene expression in plants. Since CpG islands are predicted targets for methylation, it would be good to examine their relevance using global analysis approach. There is a positive correlation between DNA methylation and gene expression under stress in rice (Rajkumar et al., Reference Rajkumar, Shankar, Garg and Jain2020), which suggests the relevance of DNA methylation in abiotic stress responses. This study provided only a lead and indicated the need for exploring the importance of CpG islands in the promoter region in regulating DRGs especially under severe stress. The correlation between the methylation status of the CpG islands and gene expression is to be established using contrasting rice genotypes under varied soil moisture regimes. The technique used in this study provided general information on genome-wide methylation, but site-specific methylation using whole-genome bisulfite sequencing and a global transcriptome analysis is needed to establish a strong relationship between DNA methylation and gene expression under stressful conditions.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1479262120000234.
Acknowledgement
Sapna Harihar would like to thank the Indian Council of Agricultural Research (ICAR) for the award of Senior Research Fellowship (SRF). This project is partially supported by the Directorate of Research, University of Agricultural Sciences, Bengaluru (No.C/B&R/C5-6815/94/2017-18) and ICAR, New Delhi.