Introduction
The requirement for alternative organ sources to supplement severe shortages of human donor organs has led to the exploration of pig-to-human xenotransplantation (Cozzi et al., Reference Cozzi, Bosio, Seveso, Vadori and Ancona2006; Yang & Sykes, Reference Yang and Sykes2007). However, the most profound obstacle to xenotransplantation is the immunologic rejection of the organ graft. The hyperacute rejection (HAR) represents the initial immunologic barrier that results in irreversible graft damage and loss within minutes-to-hours following graft reperfusion (Pino-Chavez, Reference Pino-Chavez2001). At the time of transplantation, the presence of xenoreactive natural antibodies (XNA) within the recipient triggers the HAR by activating the complement cascade, mediating rapid and irreversible destruction of the transplanted organs (Van den Bogaerde & White, Reference Van Den Bogaerde and White1997; Bach, Reference Bach1998).
Complement regulatory proteins (CRP), such as human decay-accelerating factor, hCD46 (also known as membrane cofactor protein), and hCD59, down-regulate the complement activation cascade to protect cells from damage by autologous complement (Liszewski et al., Reference Liszewski, Farries, Lublin, Rooney and Atkinson1996). Hence, transgenic pigs expressing various CRP could prevent the HAR. The transplantation of organs from these transgenic pigs has resulted in the inhibition of the complement activation cascade and reduction of the HAR (McCurry et al. Reference McCurry, Kooyman, Alvarado, Cotterell, Martin, Logan and Platt1995; Chen et al., Reference Chen, Naficy, Logan, Diamond and Adams1999; Schuurman et al., Reference Schuurman, Pino-Chavez, Phillips, Thomas, White and Cozzi2002).
Direct microinjection of recombinant DNA into the pronucleus of a zygote has been commonly used to produce transgenic animals, and especially in mice, has provided a powerful tool for the study of the regulation of gene expression in mammalian development (Gurdon et al., Reference Gordon, Scangos, Plotkin, Barbosa and Ruddle1980). Although pronuclear microinjection has been used for years to produce transgenic pigs for xenotransplantation, variable transgene expression patterns and uncertain transmission through the germ line have precluded the widespread application of this technology (Niemann, Reference Niemann2001; Piedrahita & Mir, Reference Piedrahita and Mir2004). This inefficiency may be resolved by the use of somatic cell nuclear transfer (NT) since this procedure involves selection of transgenic cells prior to the production of embryos (Niemann, Reference Niemann2001; Macháty et al., Reference Macháty, Bondioli, Ramsoondar and Fodor2002; Piedrahita & Mir, Reference Piedrahita and Mir2004). Several cloned transgenic pigs for xenotransplantation have been generated by NT using somatic cells with genetic modification (Dai et al., Reference Dai, Vaught, Boone, Chen, Phelps, Ball, Monahan, Jobst, McCreath, Lamborn, Cowell-Lucero, Wells, Colman, Polejaeva and Ayares2002; Lai et al., Reference Lai, Kolber-Simonds, Park, Cheong, Greenstein, Im, Samuel, Bonk, Rieke, Day, Murphy, Carter, Hawley and Prather2002; Ramsoondar et al., Reference Ramsoondar, Macháty, Costa, Williams, Fodor and Bondioli2003; Fujimura et al., Reference Fujimura, Kurome, Murakami, Takahagi, Matsunami, Shimanuki, Suzuki, Miyagawa, Shirakura, Shigehisa and Nagashima2004; Harrison et al., Reference Harrison, Boquest, Grupen, Faast, Guildolin, Giannakis, Crocker, McIlfatrick, Ashman, Wengle, Lyons, Tolstoshev, Cowan, Robins, O'Connell, D'Apice and Nottle2004; Takahagi et al., Reference Takahagi, Fujimura, Miyagawa, Nagashima, Shigehisa, Shirakura and Murakami2005; Nottle et al., Reference Nottle, Beebe, Harrison, McIlfatrick, Ashman, O'Connell, Salvaris, Fisicaro, Pommey, Cowan and d'Apice2007).
Embryonic germ (EG) cells are undifferentiated stem cells derived from primordial germ cells (PGC) and share morphologic, biochemical, immunologic, and developmental properties with embryonic stem (ES) cells (Matsui et al., Reference Matsui, Zsebo and Hogan1992; Resnick et al., Reference Resnick, Bixter, Cheng and Donovan1992; Labosky et al., Reference Labosky, Barlow and Hogan1994). Undifferentiated porcine EG cells that demonstrate capacities of both in vitro and in vivo differentiation have also been established (Shim et al., Reference Shim, Gutierrez-Adan, Chen, BonDurant, Behboodi and Anderson1997; Piedrahita et al., Reference Piedrahita, Moore, Oetama, Lee, Scales, Ramsoondar, Bazer and Ott1998). Unlike somatic cells that have a limited lifespan, EG cells can be cultured indefinitely in an undifferentiated state. Providing an abundance of pluripotent stem cells that can be genetically manipulated by conventional recombinant DNA techniques may enable stable genetic mutations to be established and maintained. If porcine EG cells are used as a source of donor nuclei in NT, it would be particularly advantageous in producing transgenic animals. Recently, the enhanced green fluorescent protein (EGFP) gene was efficiently introduced into porcine EG cells (Rui et al., Reference Rui, Qiu, Hu and Fan2006; Ahn et al., Reference Ahn, Won, Heo, Kang, Yang and Shim2007), and subsequent NT of these cells produced transgenic clone embryos with high efficiency, compared with conventional somatic cell NT (Ahn et al., Reference Ahn, Won, Heo, Kang, Yang and Shim2007).
Not only is the production of transgenic pigs labour-intensive, time-consuming, and costly, but the usefulness of such pigs in transplantation to humans is unpredictable. Hence, in vitro assessment of the HAR to predict the effectiveness of a transgenic approach would be important. The effect of transgene expression on reduction of the HAR can indirectly be assessed by treatment of the cells with human serum, including XNA and complement. Inhibition of human serum-mediated cytolysis in porcine embryonic fibroblast cells expressing various CRP has been reported (Huang et al., Reference Huang, Gou, Zhen, Jiang, Mao, Li, Chen and Cai2001; Lee et al., Reference Lee, Lee, Nahm, Jeon, Hwang, Paik and Rho2006). Using a similar approach, the resistance of transgenic porcine EG cells against HAR may be analyzed in vitro. Since porcine EG cells can differentiate into various cell types in appropriate culture conditions (Shim et al., Reference Shim, Gutierrez-Adan, Chen, BonDurant, Behboodi and Anderson1997; Piedrahita et al., Reference Piedrahita, Moore, Oetama, Lee, Scales, Ramsoondar, Bazer and Ott1998), differentiation of transgenic EG cells into the cells of interest, followed by serum-mediated cytolytic analysis, may provide an appropriate in vitro model system to predict the outcome of xenotransplantation prior to the actual production of transgenic pigs.
The present study was conducted to assess the reduction of the HAR in porcine EG cells following introduction of the hCD46 gene and subsequently to produce hCD46-transgenic NT embryos. The results of the study will contribute to efficiently producing transgenic pigs for xenotransplantation by providing undifferentiated nuclear donor cells with predictable efficacy in the inhibition of the HAR prior to NT.
Materials and Methods
In vitro maturation of porcine oocytes
Porcine ovaries were collected from prepubertal gilts at a local slaughterhouse and transported to the laboratory in a warm box (25–30 °C) within 2 h. Follicular fluid and cumulus–oocyte complexes (COC) from follicles 5 to 6 mm in diameter were aspirated using an 18-gauge needle attached to a 10 ml disposable syringe. Compact COC were selected and washed six times in HEPES-buffered tissue culture medium (TCM)-199 (Gibco BRL). The in vitro maturation (IVM) medium was modified TCM-199 (Gibco BRL) supplemented with 10 ng/ml epidermal growth factor (Sigma), 10 IU/ml pregnant mare serum gonadotropin (PMSG; Intervet), 10 IU/ml human chorionic gonadotropin (hCG; Intervet) and 10% (v/v) porcine follicular fluid. A group of 50 COC was cultured in 500 μl of IVM medium at 39 °C in a humidified atmosphere of 5% CO2 and 95% air. After culturing for 22 h, the COC were transferred to PMSG- and hCG-free IVM medium and cultured for another 20 h. At the end of the maturation, oocytes were freed from cumulus cells by repeated pipetting in the IVM medium containing 0.5 mg/ml hyaluronidase (Sigma) for 1 min.
Isolation and culture of porcine EG cells
Porcine EG cells were isolated from PGC of day 23 embryos collected from Hampshire × Yorkshire crossbred gilts, as described previously (Shim et al., Reference Shim, Gutierrez-Adan, Chen, BonDurant, Behboodi and Anderson1997). Briefly, the dorsal mesentery was removed from the embryos and placed in 0.02% EDTA solution (Sigma) for 20 min. Primordial germ cells were released from the dorsal mesentery by gentle pressing and pricking the tissue using fine forceps and collected by centrifugation at 800 g for 5 min. Harvested PGC were cultured in Dulbecco's modified Eagle medium (DMEM; Gibco BRL), now designated PGC culture medium, that contained 15% ES-qualified FBS (Gibco BRL), 1 mm l-glutamine, 0.1 M MEM non-essential amino acids, 10 μM 2-mercaptoethanol, 100 units/ml penicillin, 0.5 mg/ml streptomycin, and 1000 units/ml murine leukaemia inhibitory factor (Chemicon), on inactivated STO feeder cells prepared by treatment with 10 μg/ml mitomycin C (Sigma) for 2 h. Approximately 30,000 PGC were seeded per well of a 96-well plate (Falcon) containing feeder cells. The resulting EG cell colonies from the PGC culture were disaggregated by incubation in 0.25% trypsin–EDTA for 10–15 min and subcultured onto fresh feeder cells in a 4-well multidish (Nunclon) approximately every 5–7 days. All cultures were maintained at 39 °C in a 5% CO2, 95% air mixture with the culture medium changed every other day.
Feeder-free culture of porcine EG cells
The mixture of trypsinized EG and feeder cells were cultured for 15 min onto 0.1 % gelatin-coated plate until the fibroblasts were attached on the dish while most of the EG cells were floating. A pure population of EG cells was microscopically confirmed based on size since the individual EG cells were 5–15 μm in diameter, approximately one-third the size of a STO feeder cell (Shim et al., Reference Shim, Gutierrez-Adan, Chen, BonDurant, Behboodi and Anderson1997). Then, the cells in the supernatant were collected and washed by centrifugation at 800 g for 5 min, and transferred onto a 0.1 % gelatin-coated plate. EG cells were grown continuously in PGC culture medium in a humidified atmosphere of 5% CO2 in 95% air. After 4–5 additional passages, the feeder-free EG cells were used for transfection.
Transgenesis of porcine EG cells
An approximately 1.2 kb fragment of hCD46 cDNA containing the full coding region was obtained by a reverse transcription polymerase chain reaction (RT-PCR) from the total mRNA of human fibroblast cells, as described previously (Huang et al., Reference Huang, Gou, Zhen, Jiang, Mao, Li, Chen and Cai2001). A selectable neor cassette, as well as hCD46 cDNA under the regulation of a cytomegalovirus (CMV) promoter, was cloned into pTargeT vector (Promega). The transgene construct was amplified in DH5α competent cells, and the DNA was isolated using a Maxiprep kit (Promega), according to the manufacturer's protocol. To increase the efficiency of transgene integration into the genome of the EG cells, the vector was linearized by Bgl II digestion, resulting in a 6.8 kb DNA fragment containing the genes encoding hCD46 and neor under the regulation of separate promoters, as shown in Fig. 1

Figure 1 A diagram of the hCD46 transgene construct.
A day before transfection, feeder-free porcine EG cells were grown until 50% confluency per 100 mm tissue culture dish. Transfection was performed using Effectene (Qiagen), according to the manufacturer's protocol. Beginning 24 h after the transfection, transgenic cells were selected with EG cell culture medium containing 500 μg/ml of G418 (Gibco BRL) for 7–10 days. After the antibiotic selection, G418-resistant colonies were isolated and transferred on gelatin-coated dishes. Transgenic cells were continuously cultured at 39 °C in a humidified atmosphere containing 5% CO2 and 95% air. Transgenesis was confirmed by amplification of the hCD46 gene from genomic DNA samples of transfected EG cells using a polymerase chain reaction (PCR).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was prepared from transfected porcine EG cells using an Easy-Spin total RNA extraction kit (Intron Biotechnology), and aliquots of 5 μg of total RNA were used for cDNA synthesis using a SuperScript III first-strand synthesis system with oligo dT (Invitrogen), according to the manufacturer's protocol. Synthesized cDNA were amplified by Taq DNA polymerase (TaKara Korea Biomedical). Thirty-five cycles of PCR amplification were performed as follows: denaturation at 94 °C for 1 min, annealing at 54 °C for 1 min, and extension at 72 °C for 1 min. The sequences of the upstream and downstream primer pairs and lengths of PCR products were as follows: hCD46 (5′-TCAGTAGCAATTTGGA-GCGG-3′, 5′-CACTCTGGAGCAGCACGACT-3′ and 233 bp, respectively) and β-actin (5′-TACCTCATG-AAGATCCTC-3′, 5′-ATCTCCTTCTGCATCCTGTC-3′ and 391 bp, respectively).
Complement-mediated cytolytic assay
Transfected and non-transfected porcine EG cells were plated at 2 × 105 cells per well of a gelatin-coated 96-well multidish 24 h prior to assay. Then, the cells were washed with serum-free DMEM and cultured in the medium in which 15% FBS was replaced with AB-type normal human serum (Sigma). After further culture of the cells for 24 h, the number of surviving cells was counted after staining with 0.4% trypan blue (Sigma).
In addition, the mitochondrial activity of the transfected and non-transfected EG cells after human serum treatment was measured by a WST-1 cell proliferation assay system (Roche) using the manufacturer's protocol. Briefly, transfected and non-transfected porcine EG cells were plated at 2 × 105 cells per well of a gelatin-coated 96-well multidish 24 h prior to assay. Then, the cells were washed with serum-free DMEM and exposed to increasing concentrations of AB-type normal human serum for 4 h at 39°C. Cell viability was measured based on the cleavage of the tetrazolium salt WST-1 by mitochondrial dehydrogenases in viable cells relative to the no serum added condition using colorimetric detection with a microplate reader (Bio-Rad Laboratories).
Nuclear transfer
At 40–42 h after the onset of IVM, oocytes were enucleated with a 20 μm (internal diameter) glass pipette by aspirating the first polar body and the second metaphase plate with a small volume of surrounding cytoplasm in HEPES-buffered TCM-199 supplemented with 0.4% bovine serum albumin (BSA; Sigma) and 7.5 mg/ml cytochalasin B (Sigma). After enucleation, the oocytes were stained with 5 mg/ml bisbenzimide (Hoechst 33342; Sigma) for 10 min and observed under a Nikon TE-300 inverted microscope equipped with epifluorescence (Nikon Instrument). Oocytes containing DNA materials were excluded from subsequent experiments. Transfected EG cells were trypsinized into single cells, and transferred into the perivitelline space of enucleated oocytes. The resulting couplets were equilibrated for 1 min in0.28 M mannitol solution containing 0.5 mM HEPES, 0.05 mM CaCl2, and 0.1 mM MgCl2 in a chamber containing two electrodes. Then, the couplets were fused with a double DC pulse (1.5 kV/cm for 40 μs) using a BTX Electro-Cell Manipulator 2001 (Gentronics). Following electrical stimulation, the reconstructed oocytes were washed three times with NCSU 23 supplemented with 4 mg/ml fatty acid-free BSA (Sigma) and cultured in the same medium containing 7.5 mg/ml cytochalasin B for 3 h to suppress extrusion of the second polar body. Next, the reconstructed oocytes were cultured for 4 days in NCSU 23 containing 4 mg/ml fatty acid-free BSA and transferred to NCSU 23 containing 10% FBS and cultured for another 3 days. All NT embryos were cultured at 39 °C in a humidified atmosphere containing 5% CO2 in 95% air. The rate of in vitro development of NT embryos derived from hCD46-transgenic EG cells was monitored, and the embryos were analyzed for transgene hCD46 by PCR.
Detection of the transgene in EG cells and NT embryos
Transfected EG cells and blastocyst-stage NT embryos were subjected to analysis for the transgene hCD46. Genomic DNA from EG cells was prepared using a DNeasy Tissue kit (Qiagen), according to the manufacturer's protocol. In NT embryos, blastocysts were washed twice in phosphate-buffered saline, and individual blastocysts were separately transferred into 0.2 ml PCR tubes, each containing 5 μl of embryo lysis solution (pH 8.6) composed of 20 mM Tris–HCl, 0.9% Tween 20 (Sigma), 0.9% IGEPAL CA-630 (Sigma) and 400 μg/ml protease K (Sigma). Prior to PCR, embryos were incubated at 55 °C for 30 min, and the protease was inactivated by a 15 min incubation at 94 °C. For both EG cells and NT embryos, the PCR was performed using PerFect PCR premix (TaKara Korea Biomedical). Amplification of the hCD46 gene was carried out using forward 5′-CGATTAAGTTGAGTTACGCC-3′ and reverse 5′-ACGCCAAGTTATTTAGGTGA-3′ primers that gave rise to a 1369-bp fragment. Thirty cycles of PCR amplification were performed under the following conditions: denaturation at 94 °C for 1 min, annealing at 50 °C for 1 min, and extension at 72 °C for 2 min.
Statistical analysis
For the complement-mediated cytolytic assay, each experiment was performed in triplicate. Data on the cell counts and optical density (OD) values from transgenic and non-transgenic EG cells were subjected to Student's t-test. Differences of p < 0.05 were considered significant.
Results
Transgenesis of EG cells
The complement regulatory protein hCD46 gene under the regulation of a CMV promoter was introduced into porcine EG cells. Transfection of feeder-free EG cells using polycationonic lipid and subsequent selection of cells with antibiotics resulted in hCD46-transgenic EG cells. Transgenesis of EG cells was verified by PCR, as shown in Fig. 2. All three replicates of transfection appeared to be successful. In RT-PCR analysis, as represented in Fig. 3, hCD46 expression was detected from porcine EG cells.

Figure 2 PCR screening of hCD46-transfected porcine EG cells. M, size marker; 1–3, three replicates of transfection of porcine EG cells with the hCD46 transgene construct; V, vector containing the hCD46 gene; N, non-transfected porcine EG cells for negative control.

Figure 3 RT-PCR analysis of hCD46-transfected porcine EG cells. EG, non-transfected EG cells; Tg, hCD46-transgenic EG cells; F, porcine fibroblast cells; DW, negative control with the DNA sample replaced by distilled water.
Complement-mediated cell lysis in transgenic EG cells
To test the resistance of hCD46-transgenic EG cells against human XNA and complement, transgenic and non-transgenic porcine EG cells were treated with human serum. The cell viability assessed by trypan blue staining is shown in Fig. 4. A large number of non-transfected EG cells were stained by trypan blue, whereas hCD46-transgenic EG cells were not, indicating EG cells carrying the transgene hCD46 were protected from human complement-mediated cell lysis. In counts of viable cells, as shown in Fig. 5, the treatment of human serum did not affect the survival of hCD46-transgenic EG cells, whereas approximately one-half of the non-transfected EG cells failed to survive with the same treatment (p < 0.01). Similarly, in WST-1 analysis, hCD46-transgenic EG cells have a much higher (p < 0.01) mitochondrial activity than do the non-transgenic counterparts after the treatment of human serum (Fig. 6), indicating the protection from complement-mediated cytolysis by the introduction of transgene hCD46.

Figure 4 Representative photographs of complement-mediated cell lysis in hCD46-transgenic EG cells; ×100. A, hCD46-transfeced porcine EG cells; B, non-transfected porcine EG cells.

Figure 5 Survival of hCD46-transgenic and non-transgenic EG cells after treatment with normal human serum. **p < 0.01.

Figure 6 Mitochondrial activity of hCD46-transgenic and non-transgenic EG cells represented by optical density (OD) after treatment with normal human serum. **p < 0.01.
In vitro development and transgene detection in NT embryos
Transgenic EG cells, presumably capable of resistance against the HAR, were used as nuclear donors for subsequent transfer of the nucleus into enucleated oocytes. Among 235 reconstituted oocytes, 35 (14.9%) developed to the blastocyst stage, as shown in Table 1. Analysis of individual NT embryos by PCR indicated that 80.0% (28/35) of the embryos contained the transgene hCD46, as shown in Fig. 7. However, a few embryos from the nuclear donor cells that might have escaped from antibiotic selection were PCR negative.
Table 1 In vitro development and transgene detection in nuclear transfer embryos

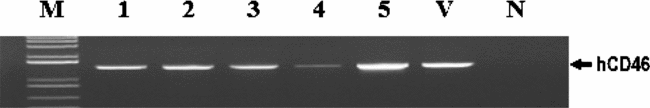
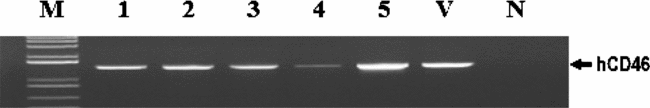
Figure 7 PCR Screening of the transgene hCD46 in nuclear transfer blastocysts. M, size marker; 1–5, porcine blastocysts obtained from nuclear transfer of hCD46 transgenic EG cells; V, vector containing the hCD46 gene; N, non-transgenic blastocysts for negative control.
Discussion
Transgenic animals can be produced by somatic cell NT. Since the procedure involves selection of transgenic nuclear donor cells prior to nuclear transfer, it may increase the efficiency in the production of transgenic animals (Niemann, Reference Niemann2001; Macháty et al., Reference Macháty, Bondioli, Ramsoondar and Fodor2002; Piedrahita & Mir, Reference Piedrahita and Mir2004). However, the rates of generating live offspring by somatic cell NT have been disappointingly low. Several factors originated from nuclear donor cells might affect the efficiency of NT. These factors include the cell cycle stage (Kasinathan et al., Reference Kasinathan, Knott, Wang, Jerry and Robl2001; Gibbons et al., Reference Gibbons, Arat, Rzucidlo, Miyoshi, Waltenburg, Respess, Venable and Stice2002; Wells et al., Reference Wells, Laible, Tucker, Miller, Oliver, Xiang, Forsyth, Berg, Cockrem, L'Huillier, Tervit and Oback2003), age (Hill et al., Reference Hill, Winger, Long, Looney, Thompson and Westhusin2000; Tian et al., Reference Tian, Xu and Yang2000; Kasinathan et al., Reference Kasinathan, Knott, Wang, Jerry and Robl2001; Liu et al., Reference Liu, Shin, Pryor, Kraemer and Westhusin2001), and culture conditions (Zakhartchenko et al., Reference Zakhartchenko, Alberio, Stojkovic, Prelle, Schernthaner, Stojkovic, Wenigerkind, Wanke, Duchler, Steinborn, Mueller and Brem1999) of the nuclear donor cells. Indeed, the type of donor cells may be one of the most important factors in determining the efficacy of NT. Although somatic cell NT has been a widely used procedure to generate cloned offspring in mammals (Galli et al., Reference Galli, Duchi, Moor and Lazzari1999; Kato et al., Reference Kato, Tani and Tsunoda2000; Ogura et al., Reference Ogura, Inoue, Ogonuki, Noguchi, Takano, Nagano, Suzuki, Lee, Ishino and Matsuda2000; Hochedlinger & Jaenisch, Reference Hochedlinger and Jaenisch2002; Miyashita et al., Reference Miyashita, Shiga, Yonai, Kaneyama, Kobayashi, Kojima, Goto, Kishi, Aso, Suzuki, Sakaguchi and Nagai2002), the transfer of stem cells with low levels of epigenetic marks may be advantageous because such cells could more easily be reprogrammable and support greater development of NT embryos compared with terminally differentiated cell types (Faast et al., Reference Faast, Harrison, Beebe, McIlfatrick, Ashman and Nottle2006). In mice, oocytes reconstructed from ES cells gave rise to an increase in the number of viable offspring compared with those from somatic cells (Wakayama et al., Reference Wakayama, Rodriguez, Perry, Yanagimachi and Mombaerts1999; Rideout et al., Reference Rideout, Wakayama, Wutz, Eggan, Jackson-Grusby, Dausman, Yanagimachi and Jaenisch2000). We have previously demonstrated that the porcine EG cell NT increased the efficiency of clone embryo production compared with conventional somatic cell NT (Ahn et al., Reference Ahn, Won, Heo, Kang, Yang and Shim2007). Porcine EG cells may be more amenable to reprogramming after reconstruction than differentiated somatic cells. Furthermore, the chromosomal stability of porcine EG cells, as previously reported (Shim et al., Reference Shim, Gutierrez-Adan, Chen, BonDurant, Behboodi and Anderson1997), may yield consistent results in NT compared with somatic cells, such as fetal fibroblasts, that often exhibit chromosomal abnormalities in long-term culture (Mir et al., Reference Mir, Tanner, Chowdhary and Piedrahita2003).
Successful acceptance of xenografts occurs after overcoming a series of obstacles, including hyperacute, acute vascular, cellular, and chronic rejection (Khalpey et al., Reference Khalpey, Koch and Platt2004; Yang & Sykes, Reference Yang and Sykes2007). Hence, production of multiple genetically modified pigs may be necessary to eliminate such hurdles (Takeuchi et al., Reference Takeuchi, Magre and Patience2005). As porcine EG cells can be maintained indefinitely in culture, use of such cells for transgenesis may facilitate ease of gene transfer and subsequent selection of transgenic cells, overcoming the current problem of using terminally differentiated somatic cells that easily become senescent during the process of gene transfer and antibiotic selection. With ease of transgenesis and maintenance of transgenic cells in culture, cloned pigs with multiple transgenes related to xenograft rejection may be produced from porcine EG cells. Recently, introduction of the exogenous EGFP gene into porcine EG cells has been demonstrated (Rui et al., Reference Rui, Qiu, Hu and Fan2006; Ahn et al., Reference Ahn, Won, Heo, Kang, Yang and Shim2007). Transfected EG cells selected by antibiotics maintained a stable expression of EGFP in culture for several months without an overt sign of differentiation (Ahn et al., Reference Ahn, Won, Heo, Kang, Yang and Shim2007). Stable maintenance of transgenic EG cells would be particularly advantageous when these cells are used for nuclear donor cells for NT to produce transgenic pigs.
In the present study, a transgene construct including the human complement regulatory protein hCD46 gene (Fig. 1) was introduced into porcine EG cells. After antibiotic selection of transfected cells, the transgene was detected from the porcine EG cells as verified by PCR (Fig. 2). The expression of hCD46 driven by a CMV promoter was analyzed by RT-PCR. As represented in Fig. 3, hCD46 was expressed in transfected EG cells, whereas no expression was detected from both fibroblast and non-transfected EG cells.
In this study, hCD46-transgenic EG cells were resistant to complement-mediated cytolysis (Fig. 4). After treatment with human serum, the survival of hCD46-transgenic EG cells was approximately two-fold (p < 0.01) that of non-transgenic controls (Fig. 5). Similarly, the mitochondrial activity of hCD46-transgenic EG cells was significantly higher (p < 0.01) than non-transgenic controls in complement-mediated cytolytic analysis (Fig. 6). Viability and mitochondrial activity of hCD46-transgenic EG cells after the treatment of human serum containing XNA and complement may reflect resistance against the HAR. Hence, transgenesis of EG cells with the hCD46 gene in this study may have prevented the EG cells from the HAR.
Application of reliable in vitro procedures to pre-evaluate the effectiveness of a transgenic approach would be beneficial in the production of transgenic pigs for xenotransplantation. In particular, porcine EG cells can be differentiated into various cell types in culture (Shim et al., Reference Shim, Gutierrez-Adan, Chen, BonDurant, Behboodi and Anderson1997). Hence, analysis of resistance against immune rejection, including the HAR, in various differentiated cell lineages following genetic modifications of EG cells may be feasible to provide an appropriate in vitro system to foresee the outcome of xenotransplantation prior to the actual production of transgenic animals.
In this study, transgenesis of EG cells was confirmed by PCR, and the introduction of the transgene hCD46 significantly enhanced the survival of EG cells from complement-mediated cytolytic attack by human serum containing XNA and complement.
These transgenic EG cells, presumably capable of overcoming the HAR, were used as nuclear donors to produce transgenic clone embryos. As presented in Table 1 and Fig. 7, analysis of individual NT embryos by PCR indicated that 80.0% of embryos contained the transgene hCD46. A small proportion of PCR-negative embryos might be due to an incomplete antibiotic selection of cells after transfection.
In conclusion, the cytolytic analysis demonstrated a reduction of the HAR in hCD46-transgenic EG cells, and the subsequent transfer of these cells into enucleated oocytes successfully produced hCD46-transgenic clone embryos. Further studies on the transfer of these transgenic clone embryos to surrogates may provide a new procedure to produce transgenic pigs for xenotransplantation.
Acknowledgements
This research was supported by the grant (108080–03–1-SB010) from Technology Development Program for Agriculture and Forestry, ministry for Food, Agriculture, Forestry and Fisheries, the grant (F104AD010002–08A0401–00220) from Korea Biotech R&D Group of Next-Generation Growth Engine Project, Ministry of Education, Science and Technology, and the grants (20050301–034–410–008–01–00 and 20070301–034–040–008–04–00) from BioGreen 21 Program, Rural Development Administration, Republic of Korea.