


Introduction
In the Developmental Origins of Health and Disease (DOHaD) framework,Reference Barker 1 , Reference Gluckman, Hanson, Cooper and Thornburg 2 many associations between the early-life environment (e.g. prenatal exposure to maternal obesity, smoking and suboptimal diet) and later health outcomes have been described.Reference Godfrey and Barker 3 – Reference Yu, Han and Zhu 6 However, the underlying molecular mechanisms are often not known. Epigenetic changes have recently emerged as a potential mechanism that may at least partially explain these associations.
What is epigenetics
The ‘epigenome’ refers to a collection of molecular mechanisms, which do not affect the DNA sequence but can affect gene regulation by changing the structure and accessibility of the DNA. Three main types of epigenetic mechanisms can be distinguished: DNA methylation, histone modifications and non-coding RNAs. These mechanisms are not independent, but rather function together as part of the larger, complex epigenetic machinery.Reference Yamada and Chong 7 Here, we focus specifically on DNA methylation, as this is currently the most studied epigenetic mark in large population studies. DNA methylation refers to the binding of a methyl group to the DNA, mainly at positions where a cytosine is located next to a guanine, a cytosine–phosphate–guanine (CpG) site.Reference Jang, Shin, Lee and Do 8 Typically (but not always), higher levels of DNA methylation at CpG sites in promoter or enhancer regions of a gene impede transcription factors from binding to the DNA, thereby inhibiting gene expression. On the contrary, methylation of CpG sites in a gene body generally leads to increased expression.Reference Jang, Shin, Lee and Do 8 As DNA methylation patterns can be passed on mitotically during cell division, they can lead to long-term alterations in gene activity and related phenotypes.Reference Ziller, Gu and Muller 9 At the same time, DNA methylation patterns can also show a considerable degree of flexibility over time, enabling cells to respond to changing internal and external inputs.
DNA methylation within the DOHaD framework
Interest in DNA methylation within the DOHaD framework stems from the fact that (a) it has been shown to respond to both genetic and environmental influences, beginning in utero, and (b) it plays a fundamental role in development.Reference Bestor, Edwards and Boulard 10 , Reference Gordon, Joo and Powell 11 Consequently, DNA methylation shows promise as a mechanism for understanding how environmental and genetic influences shape trajectories of health and disease across the lifespan.
Early life, in particular, may be a critical period for epigenetic effects on health outcomes. Extensive reprogramming of DNA methylation patterns takes place in utero.Reference Marsit 12 Thus, aberrant modifications that occur at this early stage, before tissue differentiation, could result in alterations in gene expression across many tissue types. In addition, environmental and genetic effects on organ maturation during critical periods in pregnancy and postnatally may be mediated through the epigenome. Evidence for this has come primarily from animal experimental studies, which enable careful manipulation of environmental exposures.Reference Kundakovic and Jaric 13 In humans, an early example is the work performed in the Dutch Famine Study, which has shown that exposure to famine in the first trimester of pregnancy, but not the last, is associated with DNA methylation changes at the imprinted IGF2 gene, a key modulator of fetal development.Reference Heijmans, Tobi and Stein 14 Timing-specific effects were later found for further loci, some of which also associated with cardiometabolic phenotypes, such as birth weight and low-density lipoprotein cholesterol levels.Reference Tobi, Lumey and Talens 15 , Reference Tobi, Slieker and Stein 16 Together, this work provided preliminary evidence that (very) early life may be a critical period for epigenetically mediated effects of environmental exposures on health outcomes. In addition, twin studies have been instrumental in disentangling the relative contributions of genetic, environmental and stochastic factors to DNA methylation and phenotypic variability.Reference Gordon, Joo and Powell 11 , Reference Martino, Loke and Gordon 17
Population-based cohort studies starting in early life offer an attractive platform to build on this early work and characterize associations between early-life exposures, epigenetic mechanisms and later life health. Below, we discuss the application of DNA methylation in early-life population research, including some recent findings, key challenges and recommendations for future research.
DNA methylation in early-life population-based cohort studies
In population-based studies, there is increasing interest in the role of DNA methylation changes in observational associations of adverse exposures in the preconception period, during pregnancy and in early infancy with later life common diseases. Through the development of high-throughput cost-effective arrays which analyze hundreds of thousands of DNA methylation markers across the genome, it has become possible to measure DNA methylation in (relatively) large samples. Levels of DNA methylation at these markers are then associated with determinants and outcomes of health and disease in ‘epigenome-wide association studies’ or EWASs. In addition, many long-running population-based studies have a longitudinal collection of data and biological samples in extensive biobanks. This makes it possible to study DNA methylation over time in relation to repeated measurements of exposures and outcomes and to test key mediational hypotheses.
So far, population-based research examining epigenetics within the DOHaD framework has fallen mainly into two categories as follows: (i) studies investigating associations between prenatal exposures and offspring DNA methylation patterns, for example, dietary, environmental and psychosocial exposures (Fig. 1a: b path); and (ii) studies investigating associations between offspring DNA methylation patterns in early life and later health outcomes, for example, neurodevelopment or cardiometabolic health (Fig. 1b: b path). In both cases, DNA methylation has typically been examined at birth, either in cord blood or placental tissue, given its role as a barrier and interface between maternal and fetal environments.
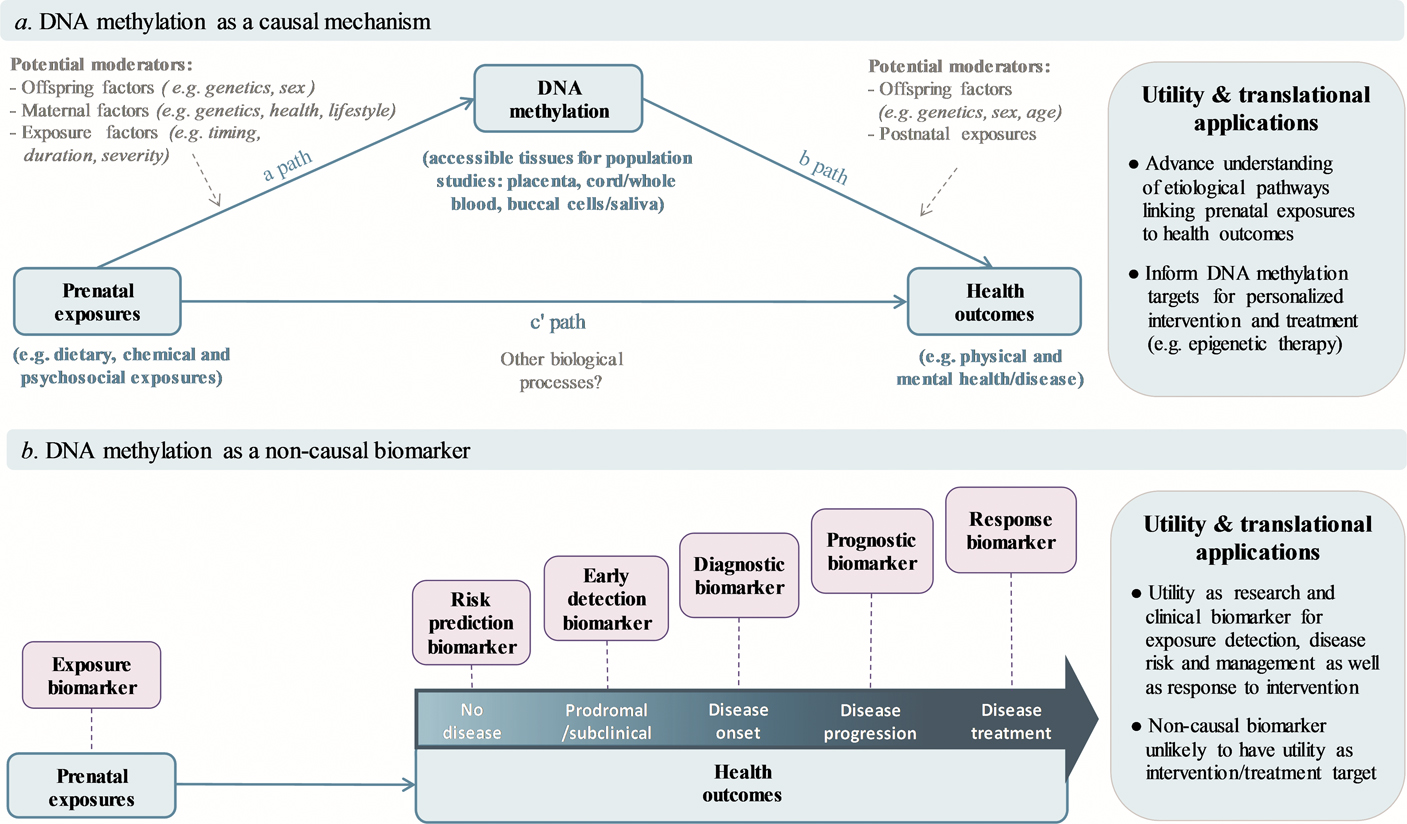
Fig. 1 (a) The causal mediation model, whereby prenatal exposures (independent variable) partly influence health outcomes in the offspring (dependent variable) via changes in DNA methylation (mediator variable). Of note, both the (a) and (b) paths are hypothesized to be moderated by genetic effects, as well as additional factors. Furthermore, DNA methylation may also mediate genetic (as well as environmental) effects. (b) The alternative non-causal model, whereby DNA methylation can serve as a biomarker of, but not a causal mechanism in, exposure-outcome associations. Note that we present here the two models that are most relevant to the Developmental Origins of Health and Disease framework; however, it is important to note that other models have also been proposed. For example, DNA methylation may function as a moderator of genetic and environmental influences on outcomes or as a mediator of genetic influences on outcomes. Moreover, stochastic changes may influence DNA methylation. More complex models are also possible (see for a more detailed discussion Ladd-Acosta et al.Reference Ladd-Acosta and Fallin 92 ).
Early-life exposures and DNA methylation
To date, the prenatal exposure that has been most robustly linked to DNA methylation changes in the offspring is maternal smoking.Reference Joubert, Felix and Yousefi 18 – Reference Rzehak, Saffery and Reischl 23 In a recent large study based on data from 13 cohorts (n=6685), Joubert et al.Reference Joubert, Felix and Yousefi 18 found that sustained smoking during pregnancy was associated with altered DNA methylation patterns across thousands of CpG sites, an effect that was still observable – albeit to a lesser extent – in older children, although the sample size was around half that of the newborn analysis. This study’s top finding was a CpG in AHRR, which has been associated with both personal and maternal smoking in multiple other studies.Reference Joubert, Haberg and Nilsen 19 , Reference Kupers, Xu and Jankipersadsing 24 , Reference Zeilinger, Kuhnel and Klopp 25 Pathway analyses found neurological and developmental pathways, which is in line with the maternal smoking-related child outcomes. Alterations in DNA methylation at birth have also been documented in relation to other chemicals, such as prenatal exposure to arsenic,Reference Green, Karagas and Punshon 26 mercuryReference Cardenas, Rifas-Shiman and Agha 27 and air pollution.Reference Gruzieva, Xu and Breton 28 , Reference Breton, Gao and Yao 29
Maternal pre-pregnancy body mass index (BMI) has also been associated with offspring DNA methylation, with findings showing associations with both lower and higher maternal BMI in a recent meta-analysis of 9340 newborns.Reference Sharp, Salas and Monnereau 30 Effects were generally small, with a median increase of 0.03% in methylation level per unit increase in maternal BMI, and strong evidence for a direct causal intra-uterine effect was lacking for most loci.Reference Sharp, Salas and Monnereau 30 Furthermore, dietary factors have been examined, with a particular focus on folate intake, due to its established role in both epigenetic regulation and fetal brain development.Reference Irwin, Pentieva and Cassidy 31 The largest of these studies, based on data from two birth cohorts (n=1988), reported associations between maternal plasma folate levels during pregnancy and differential DNA methylation in cord blood across multiple genes involved in folate biology and neurodevelopmental processes.Reference Joubert, den Dekker and Felix 32 Interestingly, this study found reduced methylation with higher levels of folate, a methyl donor, at the vast majority of CpG sites, a finding which underlines the complex associations of the one-carbon metabolism pathway with DNA methylation.Reference Joubert, den Dekker and Felix 32 In contrast, other known risk factors for poorer offspring health have not been found to be strongly associated with neonatal DNA methylation. For example, a recent meta-analysis on maternal alcohol use found no evidence for an association with cord blood DNA methylation.Reference Sharp, Arathimos and Reese 33 Similarly, a meta-analysis of prenatal maternal stress did not identify associations with DNA methylation in cord blood, although an enrichment for genes related to methyltransferase activity was identified.Reference Rijlaarsdam, Pappa and Walton 34 Besides a true null effect, the lack of a strong signal in these studies may reflect low levels of more severe forms of exposure in the general population, and relatively small sample sizes, resulting in decreased power to detect effects, or heterogeneity between the included studies in terms of population, exposure definition or confounding structure.
DNA methylation and childhood health outcomes
Compared to prenatal exposures, fewer population-based studies have examined the relationship between DNA methylation patterns and childhood health outcomes. We highlight here two important areas of progress in the field of DNA methylation in population-based studies: neurodevelopment and adiposity.
For neurodevelopmental outcomes, most of the evidence to date on this topic has been drawn from cross-sectional data in smaller, selected samples, for example, high-risk/clinical samples, although efforts to characterize these associations in the wider population are fast increasing. One such study, based on data from 537 newborns, found that epigenetic patterning of glucocorticoid genes in the placenta was prospectively associated with infant neurodevelopmental profiles.Reference Paquette, Lester and Lesseur 35 Specifically, infant membership to a reactive, poorly regulated profile associated with increased placental methylation of the NR3C1 glucocorticoid receptor gene, and decreased methylation of the HSD11B2 gene involved in limiting fetal exposure to maternal circulating cortisol. Other population studies have examined DNA methylation in relation to the risk of neurodevelopmental disorders – particularly attention-deficit hyperactivity disorder (ADHD), but also other psychiatric problems with early developmental origins, such as oppositional defiant and conduct problem behavior.Reference Barker, Walton and Cecil 36 – Reference Walton, Pingault and Cecil 39 For example, a longitudinal epigenome-wide study based on over 800 children found that DNA methylation patterns at birth (but not age 7) were prospectively associated with trajectories of ADHD symptoms from childhood to adolescence.Reference Walton, Pingault and Cecil 39 Of the 13 genome-wide significant loci identified at birth, several were annotated to genes involved in fatty acid metabolism and neural tube development. Importantly, one of the loci was annotated to a gene (ST4GAL3) that has been recently identified as the top genetic hit for ADHD in the largest genome-wide association study to date.Reference Demontis, Walters and Martin 40 Studies are also beginning to integrate brain imaging data to investigate the links between DNA methylation, brain development and neuropsychiatric outcomes. For example, a recent EWAS found that SP6 methylation at birth was prospectively associated with lower amygdala–hippocampal volume and higher psychotic symptoms in young adulthood – an association that was replicated in an independent sample of patients with schizophrenia.Reference Walton, Cecil and Suderman 41
Most of the evidence regarding DNA methylation and adiposity stems from adult studies.Reference Agha, Houseman and Kelsey 42 – Reference Wahl, Drong and Lehne 47 The largest of these identified and replicated cross-sectional associations between DNA methylation at 187 loci and BMI in over 10,000 samples. Less is known about DNA methylation and childhood adiposity. Several studies have described associations of DNA methylation with birth weight, with a limited number of CpG sites identified in more than one study.Reference Agha, Hajj and Rifas-Shiman 48 – Reference Simpkin, Suderman and Gaunt 52 No strong evidence was found for persistence of associations into later life in the two studies that explored longitudinal relationships.Reference Agha, Hajj and Rifas-Shiman 48 , Reference Simpkin, Suderman and Gaunt 52 In targeted analyses, a replicable association of RXRA methylation in umbilical cord tissue with childhood fat mass was shown.Reference Godfrey, Sheppard and Gluckman 53 Several differentially methylated regions in newborn blood, such as the promoter of the long non-coding RNA ANRIL, have been associated with childhood adiposity measures based on epigenome-wide scans.Reference Lillycrop, Murray and Cheong 54 , Reference van Dijk, Peters and Buckley 55 In cross-sectional analyses in children and adolescents, DNA methylation in several regions, including the SOCS3 locus, has been associated with adiposity measures.Reference Ali, Cerjak and Kent 56 – Reference Wang, Song and Yang 58 It remains unclear whether DNA methylation patterns are a risk factor for or consequence of adiposity. Detailed associations in the large adult EWAS show that changes in DNA methylation are mostly a consequence of obesity.Reference Wahl, Drong and Lehne 47 This is supported by smaller studies in children and individuals before and after bariatric surgery.Reference Barres, Kirchner and Rasmussen 59 – Reference Guenard, Deshaies and Cianflone 62
DNA methylation as a mediator
Despite promising findings, very few population-based studies to date have integrated prospective exposure, epigenetic and phenotypic data in order to explicitly test the role of DNA methylation as a potential biological mediator of environmental effects on health outcomes. Notable exceptions include an EWAS study based on 321 cord blood samples, which showed that, in males, DNA methylation of the PON1 gene at birth associated with prenatal mercury levels and predicted cognitive performance scores during childhood.Reference Cardenas, Rifas-Shiman and Agha 27 Using repeated measures of DNA methylation, the authors found that this effect persisted in early childhood (3–5 years) and was attenuated in mid-childhood (6–10 years). Furthermore, DNA methylation of the PON1 locus at birth was shown to be associated with gene expression levels in an independent set of cord blood samples. Another exception is a longitudinal EWAS study examining epigenetic correlates of adolescent substance use (tobacco, alcohol and cannabis use; n=244, age 14–18). This study found that neonatal DNA methylation levels in a tightly interconnected network of genes enriched for neurodevelopmental processes (n=65 epigenome-wide corrected loci) partially mediated the effect of prenatal tobacco smoking on later substance use risk in adolescence.Reference Cecil, Walton and Smith 63 Relatedly, maternal prenatal smoking has been found by other studies to associate with lower birth weight partially through alterations in cord blood DNA methylation using both targeted and epigenome-wide approaches.Reference Kupers, Xu and Jankipersadsing 24 , Reference Bouwland-Both, van Mil and Tolhoek 64
Current challenges and future directions
The field of population epigenetics is still in its infancy. Although great strides have already been taken, there is still much to discover and learn. Here we present some of the key challenges that current research in this area is facing, and how these may be addressed.
Tissue specificity
DNA methylation patterns are largely tissue-specific. In large cohort studies, blood is typically the main source of DNA collected. In some cases, there may be DNA from buccal swabs, umbilical cord or placental tissue, but other tissues are generally inaccessible for population researchers. This poses challenges, as the extent to which findings may be generalized to the target tissue of interest is often unclear.Reference Jiang, Jones and Chen 65 , Reference Davies, Volta and Pidsley 66 One way to address this limitation is to refer to publicly available datasets that show cross-tissue concordance in DNA methylation patterns, based on selected samples (e.g. comparison of blood and brain in postmortem samples or blood and adipose tissue in patients undergoing surgery), for example, http://epigenetics.iop.kcl.ac.uk/bloodbrain, and https://redgar598.shinyapps.io/BECon/.Reference Edgar, Jones, Meaney, Turecki and Kobor 67 – Reference Huang, Chu and Loucks 69 However, it is not just the concordance in DNA methylation levels between tissues, but also, and maybe more so, the variability of those DNA methylation levels in association with the exposure or outcome of interest that matters. Concordance between tissues in associations with specific phenotypes has been shown, for example, for BMI between blood, adipose and liver tissue, but overall evidence is limited.Reference Wahl, Drong and Lehne 47 , Reference Jiang, Jones and Chen 65 , Reference Stueve, Li and Shi 70 The NIH Roadmap Epigenomics Project can be used as a reference for epigenetic marks in a large number of tissues (www.roadmapepigenomics.org).Reference Kundaje and Meuleman 71
Cell type adjustment
DNA methylation patterns vary not only between, but also within tissues. In blood samples, DNA is collected from leukocytes, which represent a heterogeneous population characterized by cell type-specific DNA methylation. As such, the relative proportions of cell types from which the DNA is extracted may influence epigenetic results. Ideally, detailed measurements of leukocyte subtypes would be available for correction in all study participants, but this is often not feasible. As an alternative, the use of reference-based approaches for estimating cell type proportions has become popular, such as methods developed for whole bloodReference Houseman, Accomando and Koestler 72 , Reference Reinius, Acevedo and Joerink 73 and, more recently, cord blood.Reference Bakulski, Feinberg and Andrews 74 – Reference Gervin, Page and Aass 76 Reference-free methods are also available.Reference Kaushal, Zhang and Karmaus 77 – Reference Teschendorff and Zheng 79 These methods, however, are still under development and population-based studies with specific cell type measurements can play a pivotal role in developing and validating new analytic approaches.
Methodological considerations
DNA methylation data are multifactorial, high dimensional and inter-correlated, raising questions about how best they should be analyzed.Reference Almouzni, Altucci and Amati 80 So far, studies have varied widely in study characteristics, methodology and analytic routines, limiting comparability of findings. Furthermore, many studies have been underpowered to identify effects, especially when examining exposures or outcomes with low prevalence in the general population. To address these challenges, large collaborative efforts of prospective cohorts, such as the Pregnancy And Childhood Epigenetics (PACE) Consortium, have emerged, similar to the field of genome-wide association studies although numbers are still much smaller.Reference Felix, Joubert and Baccarelli 81 Such collaborations have shown enormous potential in increasing statistical power and optimizing the use of resources,Reference Benke, Nivard and Velders 82 – Reference Pappa, Pourcain and Benke 86 as well as allowing replication and comparison of effects between studies with potentially heterogeneous confounding structures, thus strengthening the evidence for reported associations. In addition, they are a valuable platform for standardization of study design and analysis protocols to maximize comparability and help to delineate best practices in the field. Multidisciplinary teams, bringing together basic, population and clinical scientists, will enable easier exchange of knowledge and ideas, translation between bench, population and bedside, and strengthening of scientific conclusions by bringing together multiple lines of evidence.
Role of genetics
It has been reported that a substantial proportion of the variation in DNA methylation between individuals is due to genetic variants, acting in cis or trans (mQTLs).Reference Bell, Pai and Pickrell 87 – Reference Grundberg, Meduri and Sandling 89 As such, measured DNA methylation levels may reflect genotype and mediate associations of genetic variants and health outcomes, or, if genotype drives both DNA methylation and the outcome under study, changes in DNA methylation may be an epiphenomenon of the genetic risk rather than part of the causal pathway. Furthermore, genetic factors may moderate associations between environmental exposures, DNA methylation and outcomes (Fig. 1a). Thus, genetic variability should always be considered when examining DNA methylation – ideally directly, or if unavailable or underpowered, by referring to online mQTL databases (e.g. www.mqtldb.org.)Reference Gaunt, Shihab and Hemani 88
Causal inference
DNA methylation is a dynamic process that may change under the influence of genetic and environmental factors, but also with disease states. Thus, DNA methylation may be a cause, a consequence, or an epiphenomenon of the phenotype under study (Fig. 1b). This issue of reverse causality is particularly problematic when using cross-sectional designs. While the use of prospective, longitudinal data partially addresses directionality – especially when DNA methylation is measured before the onset of disease – observational studies in general are subject to confounding. The application of advanced causal inference methods, such as two-step Mendelian randomization offers a valuable tool for probing causal pathways.Reference Relton and Davey Smith 90 For example, a recent study applied this method to support a small, causal effect of prenatal maternal B12 levels on childhood IQ levels, via DNA methylation at birth.Reference Caramaschi, Sharp and Nohr 91 The tissue specificity of DNA methylation is an additional challenge to causal inference analyses.
Determining whether DNA methylation plays a causal role in disease is important for understanding the etiology of the disease as well as potentially developing new therapies in future. Yet, even if DNA methylation changes are not causal to a disease, but rather a consequence or an epiphenomenon, they may still serve as biomarkers of the disease or its progression (Fig. 1b).Reference Ladd-Acosta and Fallin 92 In addition, if an association of DNA methylation with a phenotype is non-causal, the DNA methylation level may be used as a proxy for a specific exposure. This may be especially helpful for exposures that are difficult to measure (reliably). Previous studies have shown the potential of using DNA methylation as a biomarker in a number of these instances, including for alcohol consumption, maternal smoking during pregnancy and gestational age.Reference Bohlin, Haberg and Magnus 93 – Reference Reese, Zhao and Wu 96
Functional characterization
The buck does not stop at DNA methylation. To understand pathways leading to disease, we need to study the functional effects in terms of gene expression as well as the roles of other epigenetic marks. Effect sizes in EWAS are often small, for example, in an EWAS of maternal BMI, the largest effect estimates for the significantly associated CpG sites were an 0.1% decrease or increase in methylation level per unit increase in maternal BMI (kg/m2).Reference Sharp, Salas and Monnereau 30 For a strong exposure such as sustained maternal smoking, the maximum effect size was a 10% decrease in methylation levels for sustained smokers compared to non-smokers.Reference Joubert, Felix and Yousefi 18 It is currently unclear to what extent statistical significance parallels biological meaningfulness. In future, studies that incorporate both DNA methylation and expression data, preferably within the same population and at the same time point, can shed more light on this question of functionality. Such data are not widely available in longitudinal birth cohorts yet, with some noticeable exceptions (INMA Study, Gambia Study).Reference Guxens, Ballester and Espada 97 , Reference Moore, Fulford and Darboe 98 In addition, the ENCODE database can be used to explore whether identified DNA methylation sites are located in potential regulatory regions.Reference Barker, Walton and Cecil 36 Intensifying collaborations and expanding them to researchers in neighboring fields, such as clinical and basic researchers, will make it easier to build chains of evidence across species, across research fields and across the life course, leading to strong and innovative research.
Starting at the very beginning
Most Early-life cohorts begin in pregnancy and thus generally do not have prospective and hands-on data about the preconception and early pregnancy periods. Some preconception cohorts do exist (Southampton Women’s Survey: www.mrc.soton.ac.uk/sws,Reference Inskip, Godfrey and Robinson 99 Generation R Next Study: www.generationr.nl/next) and they, together with longitudinal birth cohorts with follow-up long enough to include the next generation, will provide valuable information on exposures in these very early life stages and their epigenetic associations. Furthermore, the vast majority of epigenetic research in birth cohorts is currently focused on maternal exposures during pregnancy. In contrast, the contribution of paternal epigenetics and exposures is currently understudied but has the potential to lend important new insights in the development of the offspring epigenome and the pathways leading from early-life exposures to child and adolescent health.
Conclusion
Population epigenetics is an emerging field with the strong potential to shed light on mechanisms underlying associations of Early-life adverse exposures and later life health. Yet, the field is still under development and poses challenges in terms of methodology and interpretation of the data. The use of multidisciplinary approaches, combining various ‘omics’ studies linking epigenetic changes to functional readouts and phenotypes, within and beyond longitudinal birth and preconception cohorts are needed to advance the field and shed further light on this complex, but highly promising, the field of research.
Acknowledgments
None.
Financial Support
J.F.F. received funding from the European Union’s Horizon 2020 research and innovation program under grant agreements No. 633595 (DynaHEALTH) and No. 733206 (LifeCycle). C.C. is supported by the Economic and Social Research Council (grant ref: ES/N001273/1).
Conflicts of Interest
None.

