Introduction
The birth of Dolly, the first cloned animal derived from a differentiated adult cell (Wilmut et al., Reference Wilmut, Schnieke, McWhir, Kind and Campbell1997), demonstrated that a fully differentiated cell can be reprogrammed to totipotential status. This innovative technology allowed consideration of the enormous potential for: (1) the production of genetically identical cattle; (2) transgenic animals for production of pharmaceutical proteins; and (3) conservation of endangered species. Fibroblasts isolated from skin tissue are frequently used as donor cells for performing bovine somatic cell nuclear transfer (SCNT) because they are among the easiest cells to culture in vitro (Freshney & Freshney, Reference Freshney and Freshney2002) and can be cryopreserved in liquid nitrogen (LN2) for future use.
Viability of frozen–thawed fibroblasts depends on several variables including cooling/thawing rates and types and concentrations of cryoprotectants that are known to affect structural integrity of plasma membranes (Muldrew et al., Reference Muldrew, Acker, Elliot, McGann, , Fuller, Lane and Benson2004). A commonly used method for freezing fibroblasts consists of adding the cryoprotectant (CPA), dimethyl sulphoxide (DMSO, 10% v/v) and freezing the cells in cryovials at a rate of ~1 °C/min in a commercially available cryo-chamber containing methanol within a low-temperature freezer (LTF; −70 to −80 °C) for ≥12 h before storage in LN2 (−196 °C; Du et al., Reference Du, Sung, Tian and Yang2002; Kragh et al., Reference Kragh, Vajta, Corydon, Purup, Bolund and Callesen2004, Reference Kragh, Du, Corydon, Purup, Bolund and Vajta2005). Development of cloned embryos to blastocyst stage and production of live calves have been obtained after reconstruction of embryos with fibroblast (Hill et al., Reference Hill, Winger, Long, Looney, Thompson and Westhusin2000), cumulus (Bhojwani et al., Reference Bhojwani, Vajta, Callesen, Roschlau, Kuwer, Becker, Alm, Torner, Kanitz and Poehland2005), granulosa (Wells et al., Reference Wells, Misica and Tervit1999) and muscle donor cells (Green et al., Reference Green, Wells and Oback2007) that were frozen in a LTF and stored in LN2. In addition, bovine fibroblast and cumulus cells have been frozen in a LTF at a cooling rate of ~2 °C/min, stored at −70 °C and used for SCNT without storage in LN2 (Kato et al., Reference Kato, Tani and Tsunoda2000; Tani et al., Reference Tani, Kato and Tsunoda2000). Development to the blastocyst stage of cloned embryos reconstructed with donor cells that had been stored at −70 °C and thawed immediately before SCNT was similar to that of cloned embryos reconstructed with donor cells that had been frozen and stored at −70 °C and then thawed and cultured before being used for SCNT (Tani et al., Reference Tani, Kato and Tsunoda2000).
To achieve the described cooling rates, it is necessary to have access to a LTF or programmable controlled-rate freezer. Such freezers are not always available in laboratories performing SCNT. However, mechanical freezers that maintain a temperature of −20 °C are almost universally available and may serve as an alternative approach for freezing donor cells for use in SCNT. McGann (Reference McGann1979) reported that hamster fibroblasts survived after being frozen at a cooling rate of 1 °C/min from −5 °C to −20 °C before storage in LN2. In addition, bovine fetal skin has been frozen in 5% DMSO and stored at −20 °C prior to derivation of fibroblast cells (Dong et al., Reference Dong, Bai, Barisanga, Matango and Suzuki2003). Rates of cleavage and development to the blastocyst stage of cloned embryos reconstructed with fibroblasts derived from frozen–thawed tissue were similar to that of cloned embryos reconstructed with fibroblasts derived from fetal skin that either were or were not frozen at −30 °C or −80 °C (Dong et al., Reference Dong, Bai, Barisanga, Matango and Suzuki2003). Although the authors of both studies reported that the cells survived after thawing and reconstructed embryos developed to the blastocyst stage, they did not evaluate the effects of cooling rate and plunging of cells from −20 °C into LN2 on subsequent cell viability and proliferation.
The CPA, a crucial ingredient for cell survival, is included in two general types of cryopreservation solutions. The first generation of cryo-solutions was designed to preserve cellular structures by inclusion of CPAs that permeate cellular membranes (Polge et al., Reference Polge, Smith and Parkes1949; Rapatz & Luyet, Reference Rapatz and Luyet1968). The second-generation of cryo-solutions contains similar components and CPAs, but has additional constituents that inhibit apoptosis, prevent free radical formation, increase buffering capacity, conserve ionic and concentration ratios, improve energy substrate availability and provide oncotic buffering (Adams et al., Reference Adams, Wang, Crane, Brown, Darlington and Ledley1995; Baust et al., Reference Baust, Buskirk and Baust2000b). Although studies have demonstrated advantages to using the second-generation of CPAs (Baust et al., Reference Baust, Baust and Van Buskirk1998, Reference Baust, Buskirk and Baust2002b; Stylianou et al., Reference Stylianou, Vowels and Hadfield2006), no studies have reported on their use with bovine fibroblasts.
Accordingly, our goals were to determine: (1) whether freezing bovine fibroblasts in a CF (−20 °C) affects viability, proliferation, apoptosis and ploidy status; (2) if cell viability is improved following freezing of fibroblasts in a second-generation of cryo-solutions; and (3) in vitro developmental competence of bovine embryos reconstructed with donor fibroblasts frozen in a CF.
Materials and Methods
Experimental design
The first experiment consisted of four treatment groups. Fibroblasts were frozen at passage 2 (P2) either by use of a: (1) LTF (~1 °C/min to −80 °C); or (2) a CF (~0.6 °C/min to −20 °C); and subsequently (3) exposed or (4) not exposed to LN2 (−196 °C) after being held at −20 °C or −80 °C for 16 to 18 h. Two to four replicates per treatment group were used to determine cell viability and apoptosis by flow cytometry.
For each of the four treatment groups, the effects of thawing in a 37 ° C or a 40 °C waterbath were examined by measuring cell proliferation and the percentage of aneuploidy. Cell proliferation was determined by counting the number of cell doublings after each passage (P3 to P12). The number of cell cycle divisions per 24 h was calculated by dividing the total cell doublings by the total number of days required for cells to reach 100% confluence. Unfrozen cells served as the control and cell doublings were counted between P1 to P7. Four replicates per treatment were performed. The incidence of aneuploidy in fibroblasts at P4 and P12 was analysed by chromosomal karyotyping for each treatment. From 43 to 56 synchronized cells were counted per group.
In the second experiment, fibroblasts at P3 were frozen either in: (1) a LTF (~1 °C/min to −80 °C); or (2) a CF (~0.6 °C/min to −20 °C); by use of either a (3) first generation cryo-solution DMEM medium supplemented with 15% fetal bovine serum (FBS; HyClone) and 10% dimethyl sulphoxide (DMSO); or (4) a second-generation cryo-solution (CryoStor CS10™, BioLife solutions). After reaching −80 °C or −20 °C cells were plunged into LN2. The numbers of live, necrotic and apoptotic cells were analysed by flow cytometry for each treatment after thawing (0 h) and after 24 h of in vitro culture. Two to four replicates were analysed.
For the third experiment, fibroblasts at P3 were thawed and cultured in vitro until they reached 100% confluence. Culture medium was replaced every other day and cells were cultured an additional 7 days. Then, cells were frozen either in a: (1) LTF (~1 °C/min to −80 °C); or (2) a CF (~0.6 °C/min to −20 °C) in CryoStor™ solution and stored in LN2. Frozen cells from each treatment were thawed and: (3) used immediately for SCNT (frozen–thawed); or (4) plated and cultured in vitro for 7 days prior to SCNT (cultured). For each treatment, the frequencies of embryo cleavage and development to the blastocyst stage were recorded. Three to five replicates of in vitro embryo development were performed per treatment.
Chemical reagents
All chemicals were obtained from Sigma-Aldrich Chemical Co., unless otherwise indicated.
Cell culture and cryopreservation of fibroblast cells
Fibroblast cell culture was generated from an ear biopsy (Uni-Punch, Premier Medicals Products) of an adult Holstein cow (Bos taurus). The skin biopsy was washed in Ca2+- and Mg2+-free Dulbecco's phosphate-buffered saline (DPBS, Gibco) and minced with a surgical blade into 1 mm2 pieces. Minced tissue was plated in a 25 cm2 (Nunc) culture flask containing 5 ml of DMEM + 10% (v/v) FBS, 6.4 mM glutamine, 0.1 mM sodium pyruvate and 50 μg/ml gentamycin at 38.5 °C in 5% CO2/air. After 7 to 10 days of incubation, monolayer outgrowths with fibroblast-like morphology at primary culture (PC) were disaggregated with 2.5 mg/ml of trypsin and passaged one to three times (P1–P3) before being frozen (frozen–thawed) or maintained in culture (fresh, control treatment).
For determining proliferation, fibroblasts were cultured in flat-sided tissue culture tubes (5.5 cm2, Nunc) at a concentration of 200 000 cells per tube until reaching 100% confluence. After each passage, the total number of cell doublings was determined by using the following formula: number of divisions = log10 (N/N0) × 3.33, where N is the total number of cells after each passage and N0 is the initial number of plated cells. Cells were counted with a Neubauer chamber and phase-contrast microscopy.
For freezing, fibroblasts at each passage were disaggregated with 2.5 mg/ml of trypsin and re-suspended in either the first generation (DMEM + 15% FBS + 10% DMSO) or second-generation cryo-solution (CryoStor CS-10™); 10% DMSO). Each precooled (4 °C) cryovial (1.2 ml, Nalgene) was filled with 500 μl of CPA solution containing ~500 000 live cells and loaded into chambers of a commercially available cryo-chamber (Mr. Frosty, Nalgene). The cryo-chambers were covered, placed either: (1) in a LTF (−80 °C); or (2) in a CF (−20 °C) and allowed to cool at ~1 °C/min or at ~0.6 °C/min, respectively, for 16 to18 h before storage of Cryovials in LN2.
Cryovials were removed from LN2 and warmed for 10 s in air at 22 °C before being submerged into a water bath at 37 °C or at 40 °C for 1 to 2 min. The CPA was removed from the cells by sequential dilution with DMEM medium. Thawed cells were analysed immediately or plated and cultured in vitro before analyses.
Cell viability and apoptosis by flow cytometry
To determine apoptosis and necrosis, frozen–thawed (0 h) and cultured cells (24 h) were centrifuged and re-suspended at ~1.5 × 106 cells/ml in 290 μl DMEM and 10 μl of 30× caspase 3-FLICA (Apoptosis Detection Kit; Immunochemistry Technologies) and cultured at 38 °C. After 2 h, cells were counterstained with 50 μg/ml of propidium iodide (PI), cultured for an additional 10 min and analysed by flow cytometry. As a positive control for apoptosis, caspase 3 was induced by incubation with 10 μM staurosporine for 12 h. Unstained cells and cells stained with only one reagent served as controls for instrument set-up.
Data from ~10 000 to 20 000 cells per sample were acquired by use of a FACScalibur (Becton Dickinson, Immunocytometry Systems) flow cytometer. Apoptotic cells fluoresced green as measured with the FL1-H detector (bandpass 530 nm) and cells with non-intact membranes fluoresced red as measured with the FL2-H detector (bandpass 585 nm). Unstained fibroblasts were considered live, cells that displayed only green fluorescence were classified as live/apoptotic and cells that displayed both red and green fluorescence were classified as necrotic/apoptotic. Cells that displayed only red fluorescence were classified as necrotic.
Chromosome analysis
Fresh (P4) and frozen–thawed (P4 and P12) fibroblasts were cultured in flat-sided tissue culture tubes (200 000 cells/tube) until reaching 50% confluence. Then, cells were incubated in DMEM medium containing 0.28 μg/ml colcemid (Gibco). After 10 h, fibroblasts were disaggregated in 2.5 mg/ml of trypsin, re-suspended in DMEM with 15% FBS and centrifuged at 1000 g for 5 min. After the supernatant was discarded, pellets were re-suspended in 5 ml of 0.075 M KCl (hypotonic solution) and incubated at 38 °C for 5 min in air. Cells were then centrifuged (1000 g for 5 min), fixed for 10 min in methanol and acetic acid (3:1) and centrifuged again at 1000 g for 10 min. The fixation/centrifugation step was repeated three times. Then, fibroblasts were placed on ice-cold wet microslides and dried in a gas flame. Fibroblast chromosomes were stained with a freshly prepared solution of 0.4% Giemsa for 10 min (Gómez et al., Reference Gómez, Pope, Lopez, Dumas, Giraldo and Dresser2006). Chromosome spreads were examined by bright-field microscopy at ×1000 to assess the chromosome composition of each fibroblast culture. Images were captured using a digital camera (D70, Olympus) and analysed with Image Pro Plus software (version 5.0.1; Media Cybernetics). Chromosome compositions were categorized as: either diploid cells containing a normal set of chromosomes (n = 60), hypoploid (n ≤ 59) or hyperploid (n ≥ 61) cells, containing less than or more than the normal diploid number of chromosomes, respectively. Chromosome spreads that were non-synchronized, scattered or overlapped were excluded from analysis.
In vitro oocyte maturation, in vitro fertilization and embryo culture
Bovine oocytes were purchased from a commercial supplier (Bomed Inc.) and delivered to New Orleans, LA by overnight express at 39 °C suspended in in vitro maturation medium (IVM). After 24 h of IVM, oocytes were co-incubated in 5% CO2, 95% air at 39 °C with 1 × 106 motile spermatozoa/ml for in vitro fertilization (IVF). Spermatozoa for IVF were obtained from frozen–thawed semen samples that were processed in TALP medium and separated by gradient-density centrifugation (Percoll®). After IVF, presumptive zygotes were vortexed to remove cumulus cells and cultured individually using the well-of-the-well (WOW) system (Vajta et al., Reference Vajta, Peura, Holm, Paldi, Greve, Trounson and Callesen2000) in 400 μl of modified synthetic oviduct fluid (SOF) medium containing 2.7 mM myoinositol, 10 μl/ml essential (BMEM, 50×) and non-essential amino acids (MEM, 100×) and 5% FBS, (SOFaa medium; Holm et al., Reference Holm, Booth, Schmidt, Greve and Callesen1999) under mineral oil (4008; Sage, BioPharma) in a humidified atmosphere of 5% CO2, 5% O2 and 90% N2 at 38.5 °C. On day 8 of culture, rates of cleavage and development to the blastocyst stage were recorded.
Embryo production by hand-made cloning (HMC) method
Hand-made cloned embryos were produced according to methods described previously (Vajta et al., Reference Vajta, Lewis, Hyttel, Thouas and Trounson2001, Reference Vajta, Maddox-Hyttel, Skou, Tecirlioglu, Peura, Lai, Murphy, Prather, Kragh and Callesen2005), with minor modifications. All manipulations were performed on a heated stage at 39 °C. Briefly, at 20 h after onset of IVM, 100 to 150 cumulus–oocyte complexes (COCs) were vortexed for 3 min in HEPES-buffered TCM199 (without FBS) containing 1 mg/ml of hyaluronidase. Cumulus-free oocytes were cultured in 0.5 μg/ml demecolcine to induce assisted enucleation. After 2 h of culture, the zona pellucida of cumulus-free oocytes was removed by 2 to 5 min exposure to 2.5 mg/ml of pronase in TCM199 without FBS. Zona-free oocytes with extrusion cones or polar bodies were placed in a 30 μl droplet of TCM199 + 20% FBS under oil and were manually bisected with an ultra-sharp splitting blade (AB Technology) while viewing with a stereomicroscope (×20). All demi-oocytes were stained with 25 μg/ml Hoechst 33342 in TCM199 + 2% FBS for 5 min and subsequent selection of those without chromatin (cytoplast) was done by epifluorescence microscopy. Exposure to ultraviolet light was restricted to 1 s. Cytoplasts were then kept in a four-well dish with 400 μl modified SOFaa medium at 39 °C before being fused.
For embryo reconstruction, one cytoplast was incubated briefly in 1 mg/ml phytohemagglutinin before being pressed gently over a single, presumably synchronized (G0/G1) fibroblast cell located in a drop of TCM199 + 2% bovine serum albumin (BSA; fatty acid free). Following attachment, the cytoplast–somatic cell pair and another single cytoplast were transferred into a 60 mm Petri dish containing 5 ml of fusion medium (0.3 M mannitol, 0.1 mM Mg2+ and 1 mg/ml polyvinyl alcohol) and positioned between two stainless-steel electrodes attached to micromanipulators (LF-101; Nepa Gene). Fusion was induced by applying a 2-s AC prepulse of 10V, 1Mhz, followed by two 35-μs DC pulses of 25 V at intervals of 2 s. Triplets (cytoplast–cell–cytoplast) were washed and cultured in 5 μl droplets of modified SOFaa medium under mineral oil and after 30 min, fusion was evaluated visually (×20).
Triplets were activated 2 to 3 h after fusion (28 h after IVM) by incubating in 5 μM Ca ionophore A23187 (in 400 μl TCM199 + 2% FBS) for 5 min at 39 °C and then in 5 μl droplets of SOFaa medium supplemented with 2 mM 6-dimethylaminopurine (6-DMAP) at 38.5 °C in 5% CO2 and 95% air under mineral oil for 4 h. As controls for the activation protocol, zona-free oocytes were parthenogenetically activated by the same procedure as described for fused NT triplets.
Following activation, reconstructed triplets and parthenogenetically activated oocytes were cultured in 400 μl of SOFaa medium in the WOW system, as described earlier. On day 8 of culture, the number of triplets and parthenogenetically activated oocytes that cleaved and developed to the morula and blastocyst stages was assessed.
Statistical analysis
Three-way ANOVA was used to analyse the data on cell proliferation, necrosis and apoptosis. The Holm–Sidak method was used to determine any differences between two means after ANOVA. The chi-squared test was used to analyse data on fusion, embryo cleavage, blastocyst development and the relative frequency distribution of chromosomal abnormalities between P4 and P12. A probability of p < 0.05 was considered to be statistically significant. ANOVA was performed by using SigmaStat version 3.1.1 (Systat Software, Inc.).
Results
Experiment 1
Flow cytometric analysis revealed that after freezing fibroblasts at ~1 °C/min in a LTF the overall percentages of live and necrotic cells (49.5 ± 4.5% vs. 50.5 ± 4.5%, respectively) were similar to that seen after fibroblasts were frozen at ~0.6 °C/min in a CF (50.6 ± 5.2% vs. 49.3 ± 5.2%, respectively); however, the values were significantly different from those of non-frozen fibroblasts (88.0 ± 0% vs. 12.0 ± 0%, respectively; p < 0.05; Table 1.)
Table 1 Flow cytometric analysis of viability and apoptosis in bovine fibroblasts frozen in two different types of freezers.

From ~15 000 to 20 000 cells were analysed in each treatment. a ,b Different superscripts within columns depict statistical significance (p < 0.05).
In addition, the percentage of live apoptotic cells was significantly higher in fibroblasts frozen in a LTF and not exposed to LN2 (18%) than those exposed to LN2 (5%) or frozen in a CF with (4%) or without (10%) exposure to LN2 (p < 0.05; Table 1). No significant differences were noted between cells frozen in a LTF and exposed to LN2 as compared with cells frozen in a CF and exposed or not to LN2. However, exposure to LN2 after cells reached freezing temperatures clearly reduced apoptosis of cells frozen in both CF (4%) and LTF (5%; Table 1).
Fibroblasts frozen at ~0.6 °C/min in a CF had lower numbers of cell doublings/24 h (0.45 ± 0.0) and required more days (9.1 ± 0.3) to reach 100% confluence at the first passage after thawing and plating, as compared with fibroblasts frozen ~1 °C/min in a LTF (0.9 ± 0.0 and 4.0 ± 0.0, respectively; p < 0.05). After reaching confluence, the total cell doublings/24 h was similar between cell treatments at each passage. Moreover, thawing temperature did not affect overall cell proliferation rates (37 °C = 0.9 ± 0.0 vs. 40 °C = 0.9 ± 0.0) of cells frozen in a CF or a LTF.
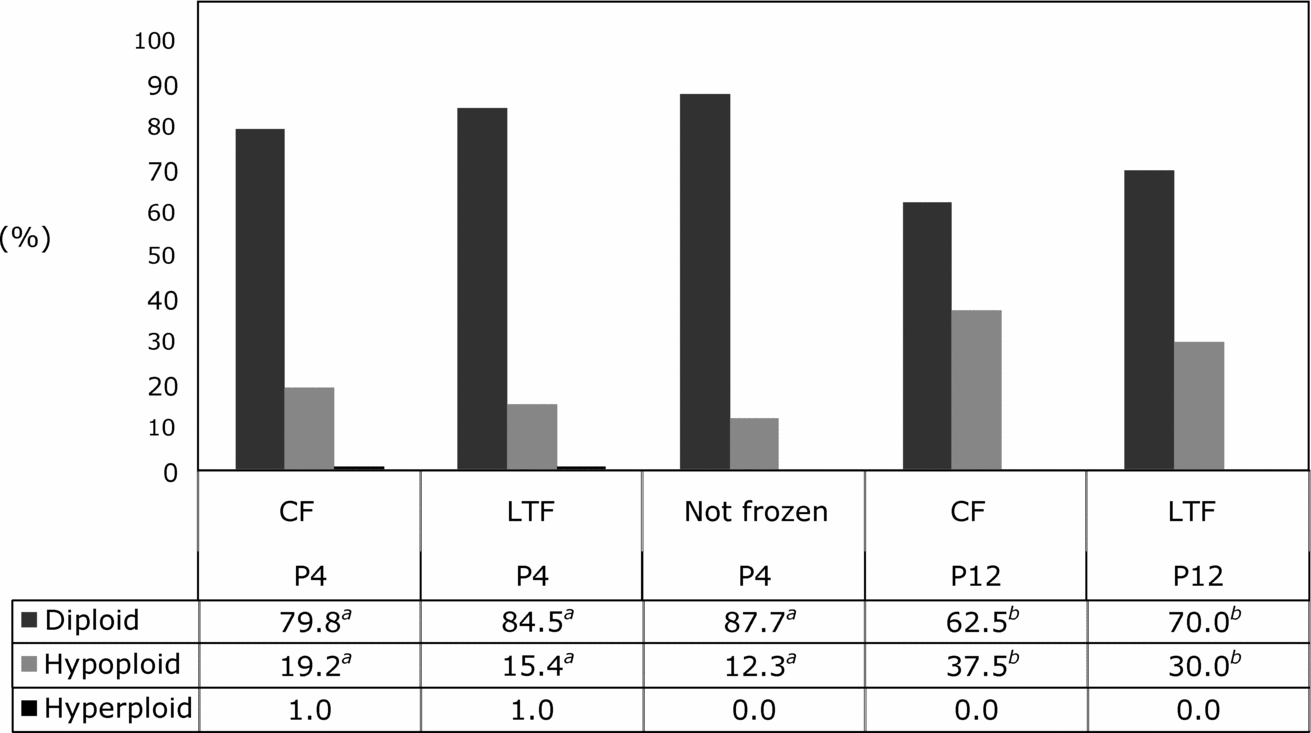
Because thawing temperature did not influence chromosomal ploidy of fibroblasts at P4 or P12, ploidy data were pooled per freezer type for statistical analysis. Similar to results observed with thawing temperature, cooling rate did not affect chromosomal ploidy of fibroblasts at P4 or P12 (Fig. 1). However, at P12 chromosomal hypoploidy of cultured fibroblasts frozen in a CF (37.5%) or in a LTF (30.0%) was higher than that of fibroblasts at P4 frozen in a CF (19.2%) or in a LTF (15.4%; p < 0.05; Fig. 1). On the other hand, chromosomal hyperploidy was not affected by duration of in vitro culture and was observed only during P4 at similar rates in both freezer types (Fig. 1).
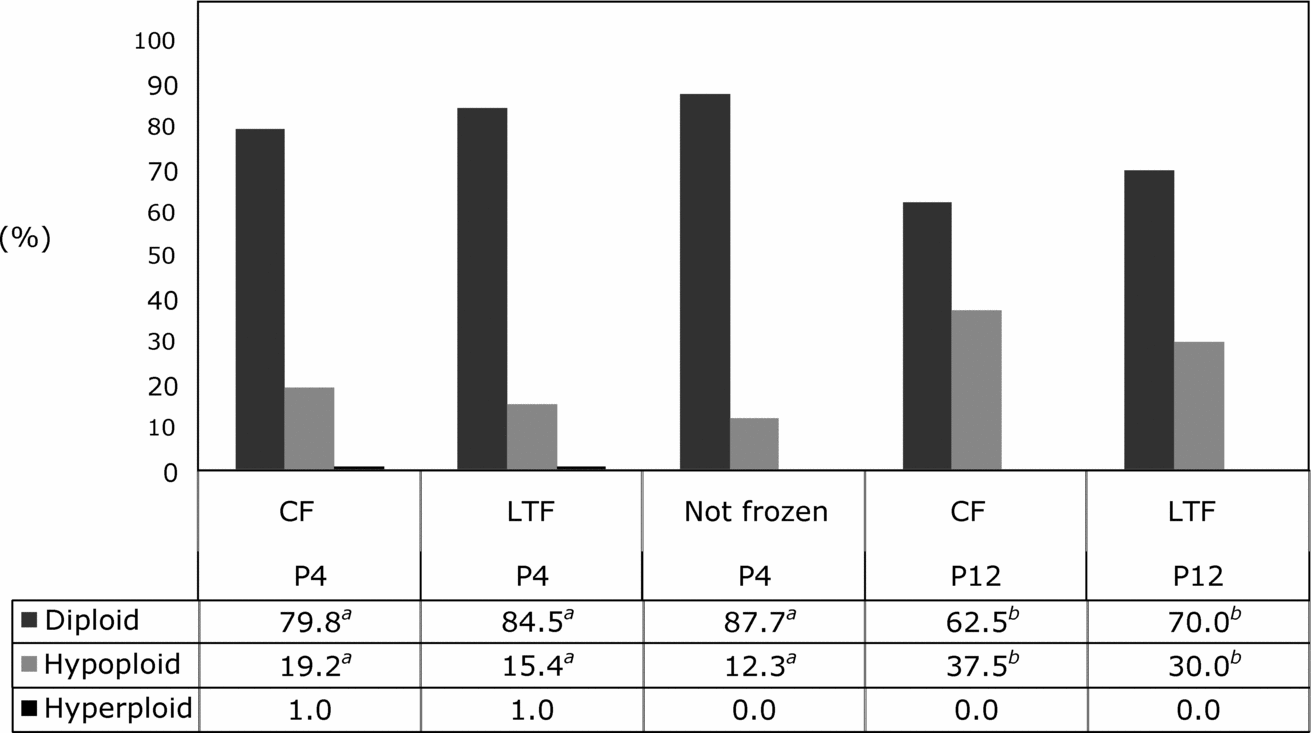
Figure 1 Chromosomal composition of bovine fibroblasts at passage 4 (P4) or 12 (P12) after freezing and thawing in a conventional or a low temperature freezer. CF, conventional freezer; LTF, low temperature freezer. a ,b Different superscripts within lines depict statistical significance (p < 0.05).
Experiment 2
The occurrence of necrotic and live/apoptotic fibroblasts after freezing in: (1) either of two cryo-solutions; and (2) a CF or a LTF (Tables 2 and 3) was determined by flow cytometry. Freezing bovine fibroblasts in a second-generation cryo-solution (CryoStor™) reduced the incidence of necrosis (29.4 ± 4.7%) at 0 h after thawing as compared with that in a first generation cryo-solution (DMEM + DMSO, 60.2 ± 5.5%; p < 0.05), but after 24 h of culture, cryo-solution type did not influence percentages of necrotic cells. However, higher percentages of necrotic cells were observed in fibroblasts frozen at ~1 °C/min in a LTF (CryoStor™ = 35 ± 6.0% vs. DMEM + DMSO = 45 ± 6.0%), than in cells frozen at ~0.6 °C/min in a CF (CryoStor™ = 20 ± 5.0% vs. DMEM + DMSO = 21 ± 5.0%; p < 0.05; Table 2). The percentage of live apoptotic cells was not influenced by the type of cryo-solution or duration of culture, but it was affected by cooling rate (CF = 1.9 ± 0.1% vs. LTF = 0.7 ± 0.1%; p < 0.05; Table 3).
Table 2 Effect of freezer and cryo-solution type on the incidence of necrosis in bovine fibroblasts at 0 and 24 h after thawing.

From ~5000 to 15000 cells were analysed in each treatment. a ,b Different superscripts within columns depict statistical significance (p < 0.05). *~0.6 °C/min to –20 °C; **~1.0 °C/min to –80 °C.
Table 3 Effect of freezer and cryo-solution type on the incidence of apoptosis in live bovine fibroblasts at 0 and 24 h after thawing.

From ~5000 to 15000 cells were analysed in each treatment. a ,b Different superscripts within columns depict statistical significance (p < 0.05) * ~0.6 °C/min to –20 °C; ** ~1.0 °C/min to −80 °C.
As in experiment 1, growth rate (doublings/24 h) after the first passage of cells frozen in a CF was lower (0.7 ± 0.0) than in subsequent passages (0.9 ± 0.0; p < 0.05). However, growth rates of cells during the first passage after freezing in a CF was not affected by the cryo-solution type (CryoStor™ = 0.75 ± 0.0 vs. DMEM + DMSO = 0.74 ± 0.0).
Experiment 3
We evaluated in vitro development of bovine embryos reconstructed with donor cells frozen in a LTF or a CF and used either immediately after thawing (frozen–thawed) or after a period of culture before HMC (cultured). Fusion rate was lower for triplets reconstructed with cells frozen in a CF and cultured before HMC (57.0%) as compared with that observed after embryo reconstruction with donor cells frozen in either a CF or a LTF and used immediately after thawing (81.0% vs. 87.0%, respectively) or frozen in a LTF and cultured before HMC (79.0%; p < 0.05; Table 4). Cleavage and development to the blastocyst stage were not affected by freezer type (i.e., cooling rate) or by using cells either immediately after thawing or after a period of culture before HMC (Table 4). However, development to the blastocyst stage after parthenogenetic activation (28.0%) or IVF (30.0%) was higher than after HMC (12.8%. p < 0.05; Table 4).
Table 4 In vitro development of IVF, parthenogenetically activated and HMC embryos reconstructed with bovine fibroblasts frozen in two freezer types and cultured or thawed immediately before HMC.

aPercentage of fused triplets (HMC), oocytes inseminated (IVF) or activated (parthenogenetic) embryos that developed in vitro. b ,c Different superscripts within columns depict statistical significance (p < 0.05).
Discussion
In the present study, we evaluated the cell viability of bovine fibroblasts frozen at a slow cooling rate of 0.6 °C/min in a −20 °C CF and measured their subsequent ability to support in vitro development after HMC. We demonstrated that viability of fibroblasts frozen in a CF was similar to that of cells frozen in a LTF; however, a higher percentage of live apoptotic cells were observed in fibroblasts frozen in a CF. Despite the lower growth rate at first passage of fibroblasts frozen in a CF, growth rate at subsequent passages and chromosomal ploidy were similar to that of fibroblasts frozen in a LTF. In addition, the second-generation cryo-solution reduced the overall incidence of necrosis in cells immediately after thawing as compared with that seen in cells frozen in the first-generation cryo-solution. Cleavage and development of cloned embryos to the blastocyst stage were similar between those reconstructed with donor cells frozen in a CF or in a LTF.
During the freezing and thawing process, cells experience modifications in their physical state, such as cell volume, membrane structure and biochemical processes, that affect survival (Mazur, Reference Mazur, Fuller, Laneg and Benson2004). Cell survival is also altered by the formation of intracellular ice, which often occurs during rapid cooling. Formation of intracellular ice can be reduced by freezing cells at a slow cooling rate (Mazur, Reference Mazur1984; Karlsson et al., Reference Karlsson, Cravalho, Rinkes, Tompkins, Yarmush and Toner1993) because the cell maintains osmotic equilibrium with its extracellular solution through dehydration (Muldrew et al., Reference Muldrew, Acker, Elliot, McGann, , Fuller, Lane and Benson2004). In our first experiment, we did not observe an effect of cooling rate on cell survival. However, in the second experiment, we observed a higher percentage of necrotic cells after fibroblasts were frozen in a LTF and a higher percentage of apoptosis in live fibroblasts frozen in a CF. It is not clear why a higher percentage of apoptosis was observed in cells frozen in a CF, but we speculate that these cells were not completely dehydrated and intracellular ice was formed after plunging into LN2. In fact, intracellular ice can induce an apoptotic-type of cell death (Baust et al., Reference Baust, Buskirk and Baust2000b).
Quantitative analysis of the movement of intracellular water in yeast and human erythrocytes frozen at a slow cooling rate (1 to 10 °C/min) from −1 °C to −30 °C indicated that the cells were dehydrated to at least 80% of their volume when they passed through the ice nucleation temperature (between −5 °C to −20 °C; Mazur, Reference Mazur1963). Although, in our study, the amount of intracellular water was not measured, we suggest that cells frozen in both type of freezers had enough time to undergo dehydration to at least 80% of their volume. Even so, the degree of cell dehydration was different between the two types of freezers. Bovine fibroblasts frozen in a LTF at a cooling rate of 1 °C/min required about 13 min to reach −20 °C and an additional 5 min and 4 h to reach −30 °C and −70 °C, respectively, whereas fibroblasts frozen in a CF at a cooling rate of 0.6 °C/min required about 42 min to cool from 0 °C down to −20 °C. Despite the longer dehydration time of fibroblasts frozen in a CF (−20 °C), there was not enough extracellular ice formed to induce cell dehydration (Pegg & Diaper, Reference Pegg, Diaper, Smit-Sibinga, Das and Meryman1990) and cells did not pass through an extra period of dehydration after reaching −20 °C. Cells with a diameter between 1 to 25 μm continue dehydrating after reaching −20 °C, but further dehydration does not occur below −26 °C (Mazur, Reference Mazur1963). Similarly, cells frozen in a LTF are exposed to lower temperatures and the probability of intracellular ice formation may be reduced by the formation of adequate extracellular ice that induces higher cell dehydration by increasing extracellular osmotic pressure (Mazur, Reference Mazur1984). Therefore, we suggest that the formation of intracellular ice in cells frozen in a CF may be due to: (1) inadequate dehydration as a consequence of incomplete extracellular ice formation during passage through the ice enucleation temperature range; and (2) insufficient cell shrinkage as a consequence of lack of exposure to temperatures below −20 °C.
Survival of hamster fibroblasts frozen at a slow cooling rate of 1 °C/min from −5 °C to −20 °C or −60 °C was increased when cells were exposed to and stored in LN2 (McGann, Reference McGann1979). Similarly, in our study, exposing bovine fibroblasts to LN2 after reaching freezer temperatures (−20 °C or −80 °C) was beneficial. Regardless of freezer type, fibroblasts exposed to LN2 had a lower incidence of apoptosis in live cells than fibroblasts that were not exposed to LN2. Possibly, plunging cells into LN2 from −20 °C or −80 °C may induce vitrification of intracellular water, thus avoiding crystal ice formation and subsequent injury that initiates the cascade of events leading to death of cells by apoptosis. Therefore, we recommend that bovine fibroblasts should be plunged and stored in LN2 after freezing in a CF.
The present study also demonstrated that freezing in a CF was detrimental to growth rate of fibroblasts. Cells frozen in a CF took more than 9 days to form a confluent monolayer at the first passage after thawing, as compared with 4 days for cells frozen in a LTF. It is possible that the higher rate of apoptosis observed in live cells frozen in a CF directly influenced the cell growth rate. In fact, apoptosis is a regulated form of cell death and is responsible for eliminating damaged cells within living tissues (Kerr et al., Reference Kerr, Wyllie and Currie1972). Caspase 3 is a major effector protease that eventually degrades crucial cellular proteins and its presence indicates that cell death is proceeding irreversibly (Thornberry & Lazebnik, Reference Thornberry and Lazebnik1998). Thus, we suggest that, although fibroblast cells survived freezing and thawing, the presence of apoptotic cells which died later during the first passage affected the growth rate, because there were fewer live intact cells that had to undergo more divisions before reaching confluence.
In vitro culture induces chromosome anomalies in bovine fibroblast cells (Giraldo et al., Reference Giraldo, Lynn, Godke and Bondioli2006). Likewise, we observed that bovine fibroblasts cultured for 38 and 40 doublings after freezing in a LTF or a CF had increased percentages of aneuploid cells as compared with earlier passages. It has been suggested that aneuploidy observed during culture is a function of chronological aging and/or proliferation history (Jacobs et al., Reference Jacobs, Court and Doll1961; Saksela & Moorhead, Reference Saksela and Moorhead1963) and that chromosomal abnormalities are present as a consequence of in vitro culture conditions. Therefore, improving in vitro culture conditions and limiting the number of passages should reduce the incidence of aneuploidy. We also suggest that cells frozen in a CF should be cultured for at least one passage before SCNT to start a normal logarithmic growth rate of live intact cells and to reduce the number of apoptotic cells.
Despite recent improvements in cell cryopreservation technology, survival rates, even within cell types, range from 20% up to 90% based on analysis performed within 1 h after thawing (Frim et al., Reference Frim, Snyder, McGann and Kruuv1978; De Loecker et al., Reference De Loecker, Koptelov, Grischenko and De Loecker1998; Baust et al., Reference Baust, Buskirk and Baust2000b). In addition, many cells die during the first 24 to 48 h of culture after thawing (Coundouris et al., Reference Coundouris, Grant, Engeset, Petrie and Hawksworth1993; Baust, Reference Baust2002). After thawing a cell, a delayed increase in transcriptional activity of caspase 3 occurs; its peak is reached at 18 h; and, its proteolytic activity is extended to 36 h post thawing (Baust, Reference Baust2002). In contrast, we did not observe an increase in caspase 3 activity during the first 24 h after thawing, but we found a reduction in cell death after 24 h of culture. Thus, the results indicate that, under our freezing conditions, molecular-based cell death after thawing occurs at a lower level at 24 h after thawing.
In our second experiment, we compared first and second-generation cryo-solutions to determine if we could increase survival rates of cells frozen in both types of freezers. Our results were similar to previous studies in that fibroblast cells frozen in CryoStor™ had a significant reduction in the incidence of necrosis at 0 h after thawing, regardless of freezer type (Baust et al., Reference Baust, Baust and Van Buskirk1998, Reference Baust, Van Buskirk and Baust2002a; Stylianou et al., Reference Stylianou, Vowels and Hadfield2006); however, the percentage of apoptosis in live cells was not affected. It is known that the addition of caspase inhibitors to cryosolutions impedes release of cytochrome c from mitochondria (Matsushita et al., Reference Matsushita, Yagi, Hardin, Cragun, Crow, Bergen, Gores and Nyberg2003) and prevents activation of the caspase pathways (Stroh et al., Reference Stroh, Cassens, Samraj, Sibrowski, Schulze-Osthoff and Los2002). Second-generation cryo-solutions contain anti-apoptotic (glutathione and α-tocopherol) and anti-oxidants agents (vitamin E) that decrease and/or prevent the detrimental effects of oxidative stress resulting from the cryopreservation process (Adams et al., Reference Adams, Wang, Crane, Brown, Darlington and Ledley1995; Baust et al., Reference Baust, Van Buskirk and Baust2000a). Although apoptosis in live cells was not reduced after freezing fibroblasts in the second-generation cryo-solution, we suggest that the protective effect on necrosis may be due partially to the presence of anti-oxidants that reduced oxidative stress. Our study demonstrated the beneficial effects of a second-generation cryo-solution on the viability of bovine fibroblasts frozen at two cooling rates.
Freezing cells at a suboptimal cooling rate can affect the nuclear reprogramming capacity of donor cells (Hayes et al., Reference Hayes, Rodriguez, Gonzalez, Falcon, Aguilar and Castro2005). In fact, cloned embryos reconstructed with donor cells frozen at a slow discontinuous cooling rate had a lower rate of development to the blastocyst stage than those reconstructed with cells frozen at a slow cooling rate in a LTF (Hayes et al., Reference Hayes, Rodriguez, Gonzalez, Falcon, Aguilar and Castro2005). However, Estrada et al. (Reference Estrada, Lee and Piedrahita2006) did not find differences in blastocyst development using two different methods to freeze pig fibroblasts for SCNT. Similarly, we observed that cleavage rate and development to the blastocyst stage were not affected by the cooling rate at which donor cells were frozen (CF vs. LTF). However, fusion percentage was lower for triplets reconstructed with cells frozen in a CF and cultured for 7 days before HMC as compared with the rate of those reconstructed with cells frozen in a CF and thawed before HMC, or triplets reconstructed with cells frozen in a LTF and thawed immediately before HMC or after culture for 7 days. It is known that the cryopreservation process induces loss of membrane integrity in frozen cells and the damage can lead to a less intimate contact between donor cell and cytoplast membrane, reducing the fusion efficiency (Hayes et al., Reference Hayes, Rodriguez, Gonzalez, Falcon, Aguilar and Castro2005). We do not know the membrane condition of fibroblasts frozen in a CF and cultured before HMC; therefore, additional studies that evaluate membrane integrity of frozen fibroblasts should be done. In addition, it remains to be determined if embryos derived from fibroblasts frozen in a CF can establish a pregnancy and produce healthy live offspring following transfer to a recipient.
Non-viable cells frozen at −80 °C without cryoprotectant or isolated from mice bodies kept frozen at −20 °C for up to 16 years can be used successfully for the production of cloned mice (Li & Mombaerts, Reference Li and Mombaerts2008; Wakayama et al., Reference Wakayama, Ohta, Hikichi, Mizutani, Iwaki, Kanagawa and Wakayama2008); however, production of live offspring relies on deriving embryonic stem cells (ESC) from cloned embryos reconstructed with non-viable cells and a second round of cloning with ESC. Such studies clearly demonstrate that viability and integrity of donor cells are crucial to the success of SCNT. The ability to cryopreserve cells successfully at different cooling rates without the use of a LTF would reduce costs and improve the opportunities for further expansion of genetic technologies. Bovine fibroblasts suspended in a second-generation cryo-solution (CryoStor™) and frozen in a CF resumed growth during in vitro culture and were used successfully as donor cells for SCNT. The present study provides documentation that conventional freezers may be used for successfully preserving bovine donor cells for SCNT, thereby broadening the scope of this biotechnology, especially for laboratories with limited resources.
Acknowledgements
This work was supported by Audubon Center for Research of Endangered Species (ACRES), USGS and, partially by a grant from the ACRES and LSU system collaborative projects. Colombian National University and Colciencias financed a fellowship and part of the residency of Liliana Chacón while she was conducting the research in New Orleans, Louisiana, USA.