


Introduction
The Equidae are important reservoir hosts for a variety of nematode parasites, some of which can cause significant morbidity or mortality if their hosts are untreated. Adult horses infected with roundworms present with a variety of clinical symptoms, including nasal discharge, coughing, anorexia, and lethal intestinal obstruction and/or rupture. Moreover, the infection of foals and yearlings can cause severe illness and even death (Morsy et al., Reference Morsy2016).
Parascaris equorum, a large roundworm infecting horses, has been found to occur in many countries, including Sudan, Egypt, Iran, the UK, Australia and China (Beasley et al., Reference Beasley, Coleman and Kotze2015; Chang et al., Reference Chang2015; Easton et al., Reference Easton2016; Ismail et al., Reference Ismail2016; Morsy et al., Reference Morsy2016; Tavassoli et al., Reference Tavassoli, Yamchi and Hajipour2016). Parascaris equorum is a well-known equine ascarid species but Parascaris univalens, which also infects horses, is often overlooked. Parascaris univalens is distributed mainly in America and Switzerland (Jabbar et al., Reference Jabbar2014; Nielsen et al., Reference Nielsen2014). Both species were first described over 130 years ago by Van Beneden (Reference Van Beneden1884). Initially, equine ascarid parasites were considered to be a single species with two substrains, namely Ascaris megalocephala bivalens and A. megalocephala univalens. Researchers then classified them into separate species, but they are notoriously difficult to distinguish morphologically (Boveri, Reference Boveri1887; Nielsen et al., Reference Nielsen2014).
In the early 1980s, cytological techniques distinguished P. equorum and P. univalens by the number of chromosomes, but this was not verified in a veterinary parasitology study (Jabbar et al., Reference Jabbar2014). The mitochondrial (mt) genome has been widely used as a genetic marker in the identification and differentiation of closely related species (Lin et al., Reference Lin2012; Liu et al., Reference Liu2012; Gao et al., Reference Gao2017). However, to date, only 14 mt genomes of horse parasitic nematodes have been reported. Although the nearly complete mtDNA sequence of P. equorum Japan isolate has been deposited in the National Center for Biotechnology Information (NCBI) database, obtaining the complete mtDNA sequence is essential to study the population genetics of Parascaris in domestic and wild horses globally. Therefore, this study aimed to determine the complete mtDNA sequence of P. equorum China isolate and to analyse the genetic relationships among different regional sources of Parascaris.
Materials and methods
Parasites and extraction of genomic DNA
Adult nematodes of P. equorum were obtained from the small intestine of a naturally infected horse in Daqing, Heilongjiang Province, China, and then washed in physiological saline. The nematodes were initially identified at the species level according to primarily morphological characteristics, using existing keys and descriptions (Taylor et al., Reference Taylor, Coop and Wall2007), then fixed in 70% (v/v) ethanol and stored at –20°C until DNA extraction. Total genomic DNA was isolated from single adult worms using sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification (Wizard® SV Genomic DNA Purification System, Promega, Madison, Wisconsin, USA). To independently verify the identity of the specimen, the internal transcribed spacer (ITS) of nuclear ribosomal DNA was amplified by polymerase chain reaction (PCR) with the universal primers NC5 (5′-GTAGGTGAACCTGCGGAAGGATCATT-3′) and NC2 (5′-TTAGTTTCTTTTCCTCCGCT-3′) and sequenced according to an established method (Gasser et al., Reference Gasser2008). The ITS (accession number: MF678787) sequence obtained was a perfect match with that of P. equorum (accession number: JN617987).
PCR amplification, sequence analyses and comparative analyses
The entire mt genome of P. equorum was amplified by PCR using 10 primers (supplementary table S1) designed from the conserved regions of Ascaris suum (HQ704901) and other Ascarididae nematodes. PCR details have been described in previous studies (Xu et al., Reference Xu2015; Zhang et al., Reference Zhang2015). Positive PCR products were sequenced at Life Technology Company (Beijing, China) using primers employed in primary amplifications.
Sequences were assembled manually and aligned against the complete mt genome sequences of A. suum (HQ704901) using the program Clustal X 1.83 (Thompson et al., Reference Thompson1997) to infer gene boundaries. Twelve protein-coding genes were identified based on comparisons with A. suum (HQ704901). The secondary structures of 22 transfer (t) RNA genes were predicted using tRNAscan-SE (Lowe & Eddy, Reference Lowe and Eddy1997) and/or manual adjustment, and ribosomal (r) RNA genes were identified by comparison with A. suum (HQ704901).
The mtDNA size, percentage of A + T content, position and length of the 12 protein-coding genes, identity of the complete mtDNA sequences, and nucleotide and amino acid sequences of the 12 protein-coding genes were compared among the four Parascaris species (P. equorum China and Japan isolates, P. univalens Switzerland and USA isolates).
Phylogenetic analyses
The amino acid sequences conceptually translated from individual genes of the mt genome of P. equorum were concatenated. Concatenated amino acid sequences predicted from the published mt genomes of 11 Ascarididae nematodes were selected for phylogenetic analyses: Ascaris lumbricoides, NC_016198; A. suum, HQ704901; Baylisascaris procyonis, NC_016200; Baylisascaris schroederi, NC_015927; Baylisascaris ailuri, NC_015925; Baylisascaris transfuga, NC_015924; P. univalens Switzerland isolate, KM067271; P. univalens USA isolate, KM216010; P. equorum Japan isolate, AP017696; Toxascaris leonina, NC_023504; and Toxocara canis NC_010690 as the outgroup. All amino acid sequences (considering all homologous characters) were aligned using MAFFT 7.122 (Katoh & Standley, Reference Katoh and Standley2013), and ambiguously aligned regions were excluded using the Gblocks online server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html), using the options for less stringent selection (Talavera & Castresana, Reference Talavera and Castresana2007). Phylogenetic analyses were conducted using two methods: Bayesian inference (BI) and maximum likelihood (ML) (Guindon & Gascuel, Reference Guindon and Gascuel2003; Ronquist & Huelsenbeck, Reference Ronquist and Huelsenbeck2003). Selected models and detailed process were as previously described in Gao et al. (Reference Gao2017). Phylograms were drawn using Tree View v. 1.65 (Page, Reference Page1996).
Results and Discussion
Mitochondrial genome organization
The complete mt genome sequence of P. equorum China isolate is 13,899 base pairs (bp) and was deposited in GenBank under accession number MF678786. It contains 12 protein-coding genes (cox1–3, nad1–6, nad4L, cytb and atp6), 22 tRNA genes and two rRNA genes (rrnS and rrnL) (table 1). All genes are transcribed in the same direction. Gene arrangement is the same as for most Ascaridida nematode genes, but is distinct from those of Ascaridiidae and Cucullanidae sequenced to date, e.g. Ascaridia galli and Cucullanus robustus (Liu et al., Reference Liu2013; Nielsen et al., Reference Nielsen2014). The nucleotide composition of P. equorum China isolate is biased towards T bases, while C is the least favoured (T = 48.63%, A = 21.62%, G = 21.59%, C = 8.16%); the A + T content of the mt genome is 70.25%, which is in accordance with mt genomes of other Ascaridida nematodes, such as A. suum and C. robustus (Park et al., Reference Park2011; Liu et al., Reference Liu2012).
Table 1. Position and nucleotide sequence length of mitochondrial genomes of Parascaris equorum and Parascaris univalens, including initiation and termination codons for protein-coding genes and their tRNA gene anticodons (starting from cox1).

PE, Parascaris equorum in this study; PEJ, Parascaris equorum Japan isolate; PUS, Parascaris univalens Switzerland isolate; PUU, Parascaris univalens USA isolate.
The total length of the 12 protein-coding sequences of P. equorum China isolate is 10,289 bp, which encodes 3419 amino acids. Of the 12 protein-coding genes, six (cox1, cox2, nad3, nad1, nad2 and nad4) use TTG as an initiation codon, three (nad5, nad4L and atp6) start with ATT, two (cytb and cox3) start with GTT, and nad6 starts with ATG. Seven genes (cox1, cox2, cox3, nad1, nad3, nad6 and cytb) use TAG as the termination codon, three (nad4L, atp6 and nad4) end with TAA, and two (nad5 and nad2) finish with incomplete terminations (T). TTG (start codons) and TAG (stop codons) were most frequently observed, which is consistent with other Ascarididae nematodes (Xie et al., Reference Xie2011; Liu et al., Reference Liu2012).
The 22 tRNA genes identified range from 54 bp (trnS1AGN) to 65 bp (trnS2UCN) in length. Prediction of their putative secondary structures (data not shown) showed that all tRNA genes have a TV-replacement loop instead of the TΨC arm and loop, except for trnS1AGN and trnS2UCN, which have the DHU loop; this is consistent with other Ascarididae nematodes, such as T. canis (Jex et al., Reference Jex2008) and A. suum (Liu et al., Reference Liu2012). rrnS and rrnL rRNA genes of P. equorum China isolate are 700 bp and 979 bp, respectively. rrnL is located between trnH and nad3, and rrnS is located between trnE and trnS2UCN. Only one non-coding region (NCR), located between trnS2UCN and trnN, was identified in the P. equorum China isolate mt genome; this is consistent with P. equorum Japan isolate, P. univalens Switzerland isolate and P. univalens USA isolate, but differs from most metazoan mtDNA sequences. For example, Rhigonema thysanophora has five NCRs (Kim et al., Reference Kim2014). The A + T content of this region of P. equorum China isolate is 80.8%, which is consistent with other Ascarididae nematodes (Liu et al., Reference Liu2012).
Comparative analyses of the mt genome of P. equorum and P. univalens
The P. equorum China isolate mt genome sequence is the smallest of the four Parascaris nematodes, being 28 bp shorter than that of P. equorum Japan isolate, and 21 bp and 451 bp shorter than P. univalens Switzerland isolate and P. univalens USA isolate, respectively. The NCR in P. equorum China isolate is 500 bp, which is also the smallest of the four Parascaris nematodes; P. equorum Japan isolate, P. univalens Switzerland isolate and P. univalens USA isolate have NCRs of 551 bp, 548 bp and 992 bp, respectively (table 1). This indicates that the size of the mt genome sequence may correlate with that of the NCR, as previously reported (Gao et al., Reference Gao2014).
Nucleotide sequences of the entire mt genomes of the four Parascaris nematodes differ by 0.1–0.9%, and total amino acid sequences of protein-coding genes differ by 0.2–2.1%. For the 12 protein-coding genes, the magnitude of nucleotide sequence variation ranges from 0 to 3.7%, and amino acid sequence differences range from 0 to 7.1% (table 2), with nad3, nad4L, and atp6 having identical protein sequences, and nad4 being the least conserved in this study.
Table 2. Identity of nucleotides and predicted amino acids for protein-coding genes in Parascaris equorum and Parascaris univalens.

PE, Parascaris equorum in this study; PEJ, Parascaris equorum Japan isolate; PUS, Parascaris univalens Switzerland isolate; PUU, Parascaris univalens USA isolate.
Phylogenetic analyses
To determine the phylogenetic relationship between P. equorum and other Ascarididae nematodes, the 12 mtDNA protein-coding genes were analysed using BI and ML methods (fig. 1). The congeneric species (Ascaris, Baylisascaris, Parascaris and Toxascaris) formed an independent branch. Nematodes of the genera Ascaris and Parascaris clustered together, further supporting the findings of Jabbar et al. (Reference Jabbar2014). Importantly, all the Parascaris species clustered based on the concatenated amino acid sequences of 12 protein-coding genes. Interestingly, P. univalens (Switzerland and USA isolates) and P. equorum (Japan and China isolates) were not classified into the same branches. A similar result was reported previously for A. suum China isolate and A. suum USA isolate (Liu et al., Reference Liu2012). The clustering of the four Parascaris species in a clade with high statistical support in the present study indicates that P. equorum and P. univalens are very closely related and may even be the same species.
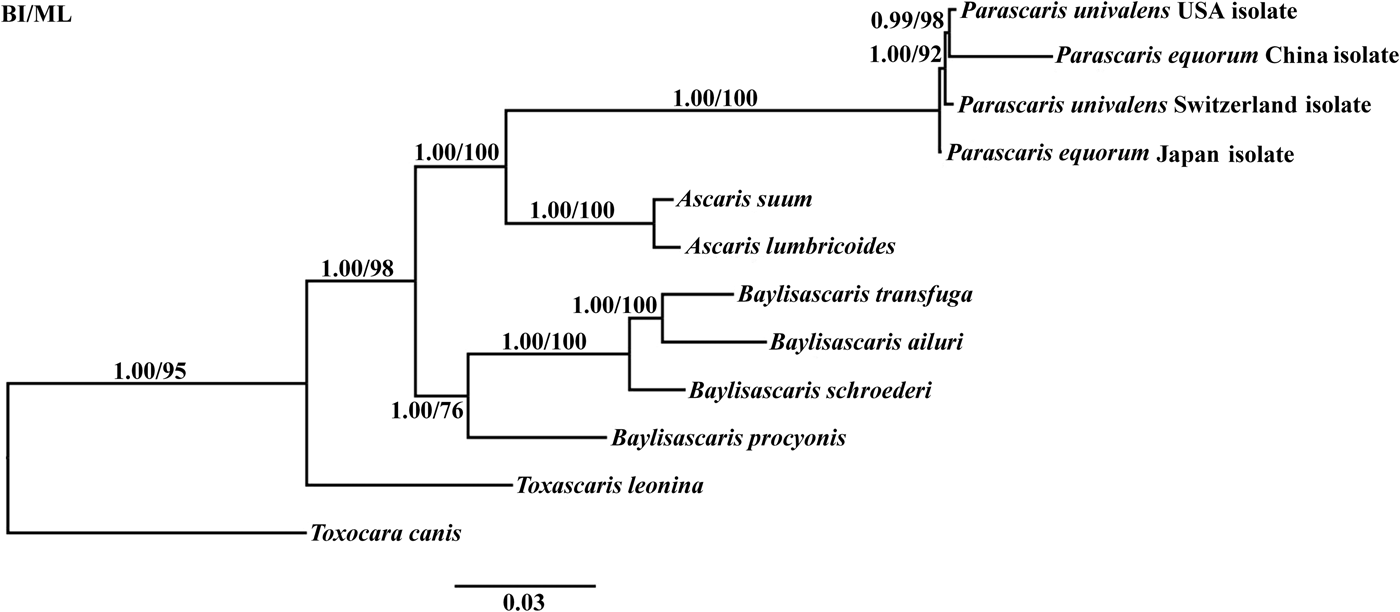
Fig. 1. Phylogenetic relationships among Parascaris species and Ascarididae nematodes based on mitochondrial sequence data. The concatenated amino acid sequences of 12 protein-coding genes were analysed by Bayesian inference (BI) and maximum likelihood (ML), using Toxocara canis as an outgroup.
In conclusion, we determined the complete mt genome sequence of P. equorum China isolate. Comparative and phylogenetic analyses of mt sequences revealed that P. equorum and P. univalens may represent the same species. The complete mt genome dataset of P. equorum extends what is known about the mt genome of parasitic nematodes.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X18000330
Acknowledgments
We thank Dr Sarah Williams of Liwen Bianji, Edanz Group China, for editing the English text of a draft of this manuscript.
Financial support
This work was supported by the National Key Research and Development Program of China (2017YFD0501306), and the Key Laboratory of Veterinary Medicine of Heilongjiang Bayi Agricultural University in Heilongjiang Provincial University (AMKL201304).
Conflict of interest
None.
Ethical standards
This study was conducted in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China, and our protocol was reviewed and approved by the Research Ethics Committee of Heilongjiang Bayi Agricultural University.





