


Introduction
The consequences of increasing globalization, due to elevated trade and passenger traffic between different continents, are becoming evident worldwide. One particularly noticeable aspect is the increased movement of species beyond their native ranges. Invasions by non-indigenous (alien, exotic) species such as pests can have spectacular impacts on their new environment. Jones & Kitching (Reference Jones, Kitching and Jones1981) define a pest as an organism that damages crops, destroys products, transmits or causes disease, is annoying or in other ways conflicts with human needs or interests. International concern about preserving biodiversity further extends the definition of a pest to a species that can either cause native species decline or alters the structure and function of natural ecosystems (Worner, Reference Worner and Schwalbe2002). More recently, pest invasions have been recognized as an important cause of loss of biodiversity, but pests as ‘vectors’ can also be considered as factors of emergence or re-emergence of viruses. For example, Begomoviruses (family: Geminiviridae), only vectored by the whitefly Bemisia tabaci, are considered as an emerging disease in many countries (Anderson et al., Reference Anderson, Cunningham, Patel, Morales, Epstein and Daszak2004). B. tabaci is vector of more than 100 begomoviruses (Jones, Reference Jones2003). Emergence of begomoviruses is considered to cause severe yield losses, but is often linked to the vector, more precisely, to the introduction of a new population (or putative species according to De Barro et al., Reference De Barro, Liu, Boykin and Dinsdale2011) of B. tabaci with an increased fitness or a wider host range, compared to the indigenous ones (Delatte et al., Reference Delatte, Duyck, Triboire, David, Becker, Bonato and Reynaud2009, Reference Delatte, Lett, Lefeuvre, Reynaud, Peterschmitt and Czosnek2007). In South America, new indigenous begomoviruses have been described after the introduction of the B species so-called B. tabaci ‘biotype B’, and declared severe epidemics; in this case, the exotic B was described as more polyphagous than the indigenous ones (Ribeiro et al., Reference Ribeiro, Ambrozevicius, Avila, Bezerra, Calegario, Fernandes, Lima, Mello, Rocha and Zerbini2003). B. tabaci was formerly thought to be a unique species composed of several well-differentiated groups, and recently those groups had been referred as species, and B. tabaci is now considered as composed of a complex of 24 cryptic species which barely interbreed and form different phylogenetic clades (Dinsdale et al., Reference Dinsdale, Cook, Riginos, Buckley and Barro2010; Xu et al., Reference Xu, De Barro and Liu2010; De Barro et al., Reference De Barro, Liu, Boykin and Dinsdale2011). Among the different clades found in the general phylogeny of Boykin et al. (Reference Boykin, Shatters, Rosell, McKenzie, Bagnall, De Barro and Frohlich2007), one is identified as comprising the ‘invasive’ biotypes/species, including B, Q and Ms. Nevertheless, only B and Q are known worldwide as invasive. The Ms, presumed to be the Indian Ocean putative species (Delatte et al., Reference Delatte, Reynaud, Granier, Thornary, Lett, Goldbach and Peterschmitt2005b), has never been considered as invasive so far. Its presumed indigenous status is supported by the presence of several monophyletic endemic begomoviruses, only vectored by B. tabaci (Delatte et al., Reference Delatte, Martin, Naze, Golbach, Reynaud, Peterschmitt and Lett2005a; Lefeuvre et al., Reference Lefeuvre, Martin, Hoareau, Naze, Delatte, Becker, Reynaud and Lett2007) and documented on some of the islands of the region (Mayotte, Madagascar and Seychelles). The biology and genetics of this putative species was studied in Réunion, together with the invasive B which recently invaded the island (Delatte et al., Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006, Reference Delatte, Duyck, Triboire, David, Becker, Bonato and Reynaud2009). In Delatte et al. (Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006), for the first time, apparently fertile hybrids (presence of hybrids after multiple generations) between B and Ms were found in the field in Réunion. Nevertheless, they were found at low frequency, with a majority of pure B or pure Ms individuals. Furthermore, no extensive sampling in the other islands was performed. To assess the Ms diversity presumed to be indigenous to this region, we infer the genetic structure and the geographical range of Ms using microsatellite data and mitochondrial DNA (cytochrome oxydase I) sequences from all the main islands of the region and samples from mainland Africa.
Materials and methods
Sample collection
Wild samples of B. tabaci were collected from most of the larger islands of the SWIO: Madagascar (2001), Réunion (2005), Mauritius (2004), Seychelles (2003), Grande Comore (2005), Anjouan (2005) and Mayotte (2005), and also from one country of mainland Africa, Tanzania (2009) (table 1, fig. 1). All samples were obtained as ethanol-preserved adults.

Fig. 1. Map of the sub-region of the southwest part of the Indian Ocean with the repartition of Bemisia tabaci B and Ms according to this study and Sseruwagi et al., 2005. Geological times are also indicated for each island, in million years (from Warren et al., 2005). Beside Seychelles and Madagascar, which are continental islands, the other islands are of volcanic origin. GC, Grande Comore; MH, Mohéli; AJ, Anjouan; YT, Mayotte; RE, Réunion; Mu, Mauritius.
Table 1. Bemisia tabaci samples repartition by country, town (locality), host plants, number of individuals tested (n) and accession number.

Microsatellite analysis
Male whiteflies are haploid, and females are diploid. Each field-captured whitefly, therefore, was sexed under a binocular microscope, and only females were used for DNA extraction due to their diploid state (see Delatte et al., Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006). A total of 882 females were analysed in this study. Nine microsatellite markers with fluorescent labels were used in this study (P5, P7, P53, P62, P11, P32, P59 (Delatte et al., Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006), Ms145 (Dalmon et al., Reference Dalmon, Halkett, Granier, Delatte and Peterschmitt2008) and Bem25 (De Barro et al., Reference De Barro, Scott, Graham, Lange and Schutze2003)). Primer sequences and methods used for DNA extraction, amplification, electrophoresis and allele scoring followed (De Barro et al., Reference De Barro, Scott, Graham, Lange and Schutze2003; Delatte et al., Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006). The genotyping phase was conducted in an ABI PRISM 3100 (©Applied Biosystem) automated sequencer.
Mitochondrial DNA
The mitochondrial cytochrome oxidase I (COI) gene, known as diagnostic for whitefly biotypes/species, was amplified and sequenced for 76 of the 882 wild SWIO individuals already typed for microsatellites, as well as for nine individuals collected in Tanzania. We used primers COI-F-C1: CATCTAATCAGCAGTGAGGCTGG and COI-R-C1: AAAAGTTAAATTTACTCCAAT. The PCR was conducted in a final volume of 25 μl with 10× PCR Optibuffer (Eurogentec), 0.2 mM dNTPs (New England Biolabs), 1.5 mM MgCl2, 400 nM of each primer, 1 unit of DAp GoldStar®, (Eurogentec) and 10 ng of insect DNA extract. The PCR programme was a cycle of 35 times: 1 min at 94°C, 30 s at 55°C and 1 min at 72°C, then a final step at 72°C for 7 min.
Data analysis
Genetic diversity within each cluster was quantified by the number of alleles per locus, the observed heterozygosity (H o) and gene diversity (H e). FREEna software was used to estimates null allele frequencies for each locus and population analysed following the Expectation Maximization (EM) algorithm (Chapuis & Estoup, Reference Chapuis and Estoup2007) with 1000 bootstrap iterations. For the two clusters separately, F st using the ENA correction described in Chapuis & Estoup (Reference Chapuis and Estoup2007) were given by FREEna; Weir & Cokerham (Reference Weir and Cockerham1984) estimates of F is within localities were calculated using Genepop 3.3 (Raymond & Rousset, Reference Raymond and Rousset1995). The null hypotheses of Hardy-Weinberg frequencies within populations, and lack of population structure, were tested with exact tests using Genepop 3.3. Allelic richness was estimated using F stat V9.3.2 (Goudet, Reference Goudet2001) based on minimum sample size of 30 diploid individuals per country for Ms populations (both countries Mauritius and Anjouan were not included in the analysis due to the smaller sample size). A hierarchical analysis of molecular variance (AMOVA) was obtained using the software Arlequin (Excoffier et al., Reference Excoffier, Laval and Schneidern2005), partitioning the genetic variance into three components: (i) within-site within-cluster, (ii) among-sites within cluster, and (iii) between clusters. Gene flow (Nm) between populations was estimated with Genepop 3.3 and the method of Barton & Slatkin (Reference Barton and Slatkin1986).
The species distribution was checked with the Structure software (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000) as described in Delatte et al. (Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006). This software differentiates mixed populations based on allele frequency at each locus. To use Structure, Hardy-Weinberg (HW) and linkage equilibrium are assumed within each group. Both hypotheses were tested a posteriori on each cluster using exact tests implemented in Genepop 3.3 (Raymond & Rousset, Reference Raymond and Rousset1995). Nevertheless, it has recently been proven that the bias linked to HWE in assignment tests, such as the ones implemented in Structure, lead only to a slight reduction in the power of the test (between 0.2 and 1.0% units) and could not affect the results (Carlsson, Reference Carlsson2008). The software TESS (Chen et al., Reference Chen, Durand, Forbes and François2007) also infers population structure, making use of geographical data. It was used for our dataset of Ms across islands excluding B. Analyses in TESS were run for 100,000 sweeps (the first 10,000 discarded as burn-in, with K ranging from 1 to 25 and 100 iterations for each value of K). Twenty percent of the 100 runs, representing the best Deviance Information Criterion, was kept. The range of K which best explained the data was inferred by plotting the average values of the DIC for each K and following the recommendations of Chen et al. (Reference Chen, Durand, Forbes and François2007). For each K, CLUMPP v1.1.2 (Jakobsson & Rosenberg, Reference Jakobsson and Rosenberg2007) was used to summarise the posterior estimates of cluster memberships of the 20 runs with the lowest DIC. We used the Large K Greedy algorithm with random input order and 1000 permutations to align the runs and the G’ pairwise matrix similarity statistics. Admixture proportions of samples and individuals were visualised using DISTRUCT v1.1 (Rosenberg, Reference Rosenberg2004). The admixture models implemented in TESS allowed us to predict expected admixture proportions on every point of the SWIO map. Each element of the matrix found represented the depth value of a particular location on the map (Chen et al., Reference Chen, Durand, Forbes and François2007). The admixture proportions resulting from TESS (K=7) were interpolated with the geographic distribution of individuals using the ‘maps’ and ‘fields’ libraries of R http://www.r-project.org/ with a universal kriging function to obtain posterior predictive maps of admixture proportions. The R script was modified to include only the SWIO region (see R Script/krigAdmixProportions). A correspondence analysis (COA) was performed using Genetix 4.01 (Belkhir et al., Reference Belkhir, Borsa, Chikhi, Raufaste and Bonhomme1996–2004) in order to visualize the major axes of genetic variation within the sample. Isolation by distance was tested within Ms populations with the correlation between genetic and geographical distance, tested by the regression of F st(1–F st)–1 on the logarithm of geographical distance (Rousset, Reference Rousset1997). A neighbour-joining tree was constructed from the Cavallis-Forza distance matrix given by Genetix 1.01 (Belkhir et al., Reference Belkhir, Borsa, Chikhi, Raufaste and Bonhomme1996–2004) and drawn in Darwin 5.0.132 (Perrier et al., Reference Perrier, Flori, Bonnot, Hamon, Seguin, Perrier and Glaszmann2003).
Sequences obtained from COI sequencing were aligned with software DNAMAN version 5.2.2 (Lynnon BioSoft, Quebec, Canada) and MEGA4 (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007). Sequences were then analysed under the module APE of R (R Development Core Team, 2004), including sequence alignment by ClustalX. The model of sequence evolution best fitting the data was then selected using PhyML. Clade support was evaluated by bootstrapping, using 2000 pseudoreplicates and performed under PhyML. Genetic distances were estimated using the method of Nei & Gojobori (Reference Nei and Gojobori1986) for synonymous and nonsynonymous substitutions, and diversity indices were calculated using the software DnaSP version 4.10 (Rozas et al., Reference Rozas, Sanchez-Delbarrio, Messeguer and Rozas2003). Genetic variation was assessed by calculating the average number of pairwise nucleotide differences among the sequences (π).
Results
A total of 882 B. tabaci were analysed, and individuals with more than 33% of missing microsatellite loci were discarded (only nine individuals were discarded from the study, two from Seychelles and seven from Madagascar). Overall, an average of 18 alleles per locus were observed for the populations of Madagascar (n=422) with an allelic richness of 9.5, five alleles for the Seychelles (n=54) with an allelic richness of 4.4, 12 alleles for Mayotte (n=113) with an allelic richness of 8.6, five for Anjouan (n=7), three for Mauritius Ms (n=5), seven for Réunion Ms (n=96) with an allelic richness of 5.7 and 12 for Grande Comore (n=75) with an allelic richness of 10.1. The highest diversity was found in Madagascar Ms populations, albeit with a higher number of samples used.
Species differentiation
The complete dataset was analysed with Structure, which identified two clusters according to the Evanno et al. (Reference Evanno, Regnaut and Goudet2005) method, including the B and Ms controls of the laboratory strains, respectively. Across our dataset, nine hybrids between B and Ms were found on Réunion, and these were discarded from the dataset. No hybrids were found on the other islands. A second check was made by sequencing the COI fragment of nine to 21 individuals randomly chosen from each species and each island. Species assessment by both techniques was similar. As a result of its useful allele size pattern, locus Ms 145 was revealed as a diagnostic locus to differentiate B from Ms. Indeed, allele sizes ranged from 170 to 200 for Ms, and from 210 to 225 for B. It has also shown utility in differentiating B from Q (Dalmon et al., Reference Dalmon, Halkett, Granier, Delatte and Peterschmitt2008).
Our results show that Ms was found on all the sampled islands of the SWIO: Seychelles (Mahé), Grande Comore, Anjouan, Mayotte, Madagascar, Réunion and Mauritius (fig. 1, table 1), whereas B was only found on Réunion and Mauritius. Furthermore, B populations were only found on crops, whereas the Ms was found on both crops and weeds in all islands where it occurred. On Réunion, Ms was found mostly on weeds (table 1).
Isolation by distance
The regression of F st(1–F st)–1 calculated across all loci on geographical distance was assessed to determine if there is a relationship between genetic distances and geographical distances. The regression was correlated with a highly significant Mantel test (Pearson r=0.883; Mantel P=0.002, with unilateral test done on 1000 permutations) for the Ms (fig. 2a).

Fig. 2. (a) Plot of geographic and genetic distances for Bemisia tabaci Ms (r2=0.71), using all the individuals of the study. (b) Neighbour-joining tree constructed on a distance matrix for the different populations of B. tabaci Ms (using microsatellite data, with Cavallis-Forza distance).
A neighbour-joining unweighted tree was constructed for the microsatellite data using the Cavallis-Forza distance matrix for the different island populations. This illustrates the large divergence between Ms populations, especially between the Seychelles population and all the others (fig. 2b). Réunion and Mauritius Ms populations are little diverged, as are Madagascar and Mayotte Ms populations.
TESS analyses implementing genetic and geographic coordinates, based on Bayesian analyses, showed the presence of seven populations (lowest DIC value) among the Ms population. The seven clusters found, plotted with a kriging function on the SWIO map, showed well-defined groups, largely reflecting the geographical distances between landmasses (fig. 3). The seven clusters are comprised of individuals from the following islands: cluster 1, Madagascar; cluster 2 Grande Comore; clusters 3, 4 and 5, Madagascar and the closest islands of the Comoros archipelago (Anjouan and Mayotte), respectively; cluster 6, Réunion and Mauritius; cluster 7, the Seychelles islands. This last analysis is congruent with the previous linear regression, showing strong genetic structure with geographic distance.

Fig. 3. Posterior predictive maps of admixture proportions as resulting from the interpolation of coancestry coefficients and the geographic distribution (restricted to the Indian Ocean region) of the seven Ms Bemisia tabaci populations obtained in TESS (each cluster corresponding to a K population found by TESS). Cluster 1, includes Madagascar; cluster 2, Grande Comore; cluster 3, 4 & 5, Madagascar, Comoros archipelago and Mayotte; cluster 6, Réunion and Mauritius; and cluster 7: Seychelles islands.
Genetic structuring
For eight loci of Ms and five of B (table 2), the average observed heterozygosity (H o) was lower than the average expected heterozygosity (H e). The average within-population heterozygote deficiencies were higher for the Ms (0.14 and –0.11, respectively; table 2). The Ms showed genetic substructure between sampling sites, as illustrated by high values of corrected F st for null alleles (table 2) and previous results. F st values were highest for the Ms. Low proportions of null alleles were detected through our dataset for B and Ms (rMs<0.17 and rB<0.03; table 2). Cases of linkage disequilibrium were detected for Ms, not for B (see Appendix 1).
Table 2. Genetic diversity of Bemisia tabaci B and Ms within and among sites of all the islands of the Indian Ocean. The observed heterozygosity (H o), the expected heterozygosity (H e) and the fixation indices (F is and F st using the ENA correction described in Chapuis & Estoup (Reference Chapuis and Estoup2007)) of Weir & Cockerham (Reference Weir and Cockerham1984) were given by the softwares Genepop and FreeNA. Hardy-Weinberg tests are indicated together with F is values. Significant P-values are presented by an *. The P-values were combined over all loci using Fisher's method. Estimate of null allele frequency (r) was also implemented.

The hierarchical AMOVA tests between species showed most variation to be within individuals within populations (66.84%) and among species (21.6%) (table 3a). The genetic differences between the B and Ms account for much more genetic variance (21.23%, F ct=0.216, P<0.001) than those among populations within the same species (11.53%, F sc=0.147, P<0.001). The differentiation among populations is present both within the B and within the Ms. However, due to low sample numbers for the B, no F-statistics are significant for the B.
Table 3. Analysis of molecular variance computed by the method of Excoffier et al. (Reference Excoffier, Laval and Schneidern2005) on samples of Bemisia tabaci B and Ms from the Indian Ocean Islands. (a) F-statistics of genetic differentiation between B and Ms biotypes and among sampling sites of B. tabaci (populations). (b) F-statistics of genetic differentiation between Ms populations of Madagascar and all the other islands of the SWIO and among sampling sites.

* stands for significant values.
The second hierarchical test for the Ms populations (between pairs of populations of the different islands) showed significant differences between all pairs analysed (table 3b). The highest F ct was found between Madagascar and Mauritius ((F ct=0.147, P<0.001), between Madagascar and Réunion (F ct=0.114, P<0.001) and between Madagascar and the Seychelles (F ct=0.182, P<0.001). The lowest F ct values were found for Madagascar and two of the populations of the Comoros archipelago (Mayotte and Grande Comore).
The matrix of Nm estimates presented in table 4 shows that the Ms populations of the Seychelles were the only ones with values less than 1 (0.62 to 0.82) for all the tested populations except with Madagascar (1.04).
Table 4. Average number of migrants (Nm)* of Bemisia tabaci Ms and B within the South Islands of the Indian Ocean.

* Nm, (1–F st)(4×F st)–1 estimation of genetic flow between pairs of populations (Genetix).
The Ms population pairs of: Madagascar/Anjouan, Mayotte/Madagascar, Mayotte/Anjouan and Grande Comore/Anjouan had the highest gene flow estimation (Nm). A high gene flow between B was also estimated between the islands of Mauritius and Réunion (8.71).
Mitochondrial DNA analysis
A total of 85 individuals was sequenced for a portion of 490 nucleotides of the mitochondrial COI gene, of which 428 nucleotides were readable and used in this study. Among those individuals, 51 belonged to the Ms, while the rest belonged to the B (table 5). Sseruwagi et al. (Reference Sseruwagi, Legg, Maruthi, Colvin, Rey and Brown2005) demonstrated the presence of B. tabaci Ms in Uganda, and the nine sequences from this study deposited in Genbank were added to our analysis (AY903521, AY903522, AY903524, AY903526, AY903530, AY903543, AY903544 AY903547 and AY903548). Overall, nucleotide diversity (π) among the Ms populations ranged from 0.00052 in Mayotte/Tanzania to 0.00584 in Uganda. The lowest diversities were recorded for the B, despite an equivalent pool of sequences studied compared to the number of localities sequenced. A phylogeny was reconstructed with those sequences and the GTR+I model was selected as the best-fitting model of DNA substitution for the data set. A lack of monophyly for island Ms populations was observed (fig. 4, table 5). However, the deepest divergence involves haplotypes from the Seychelles from all other haplotypes.

Fig. 4. Maximum likelihood analysis of the mitochondrial COI dataset for all samples used in table 5 of Bemisia tabaci. Clade support was evaluated by bootstrappings, using 2000 pseudoreplicates and performed under PhyML. Boostraps above 0.6 are indicated.
Table 5. Genetic diversities calculated from a partial mtCOI fragment of 428 nucleotides among Bemisia tabaci B and Ms.
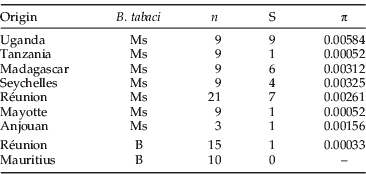
n, number of sequences used; S, the number of segregating sites; π, the nucleotide diversity.
Discussion
Three lines of evidence strongly support the action of natural processes in the divergence of the Ms within the SWIO region. Since patterns of genetic divergence in introduced species are often highly incompatible with natural processes, the patterns observed in the Ms support its status as indigenous to the SWIO region.
The first line of evidence supporting this status is that on all the islands supporting indigenous begomoviruses – Madagascar, Mayotte and the Seychelles (Delatte et al., Reference Delatte, Martin, Naze, Golbach, Reynaud, Peterschmitt and Lett2005a; Lefeuvre et al., Reference Lefeuvre, Martin, Hoareau, Naze, Delatte, Becker, Reynaud and Lett2007) – no species of B. tabaci other than Ms was found. Begomoviruses are viruses only transmitted by B. tabaci; and, on the basis of previous studies, the virus species present in the SWIO represent a distinct monophyletic group with close relationships to monopartite and bipartite African begomoviruses.
The second line of evidence is the strong correlation between nucleotide diversity of the Ms and island age and size. If introduced, one might not expect the pattern to be so clear. Within the SWIO islands, the highest diversity within the Ms was observed within the population collected on Madagascar and the Seychelles. This is consistent with the Ms populations having radiated from these islands to most of the islands. Nucleotide diversity further appears to be related to both the age and size of islands; Madagascar and the granitic Seychelles are continental blocks formed by the separation of Gondwana (Rabinowitz et al., Reference Rabinowitz, Coffin and Falvey1983), while the Mascarenes and Comoros are much more recent archipelagos with islands having formed within the last 0.13–15 Ma (Warren et al., Reference Warren, Bermingham, Prys-Jones and Thebaud2005). A strong correlation (R2=0.75) is obtained between the nucleotide diversity of each group of Ms from the different islands of the SWIO and the age of these islands (see Appendix 2). Furthermore, despite a lack of monophyly for island populations, the COI phylogeny demonstrated that the deepest splits in the Ms concern the divergence of one haplotype only found on the oldest islands from the principal grouping (Seychelles). In other words, based on COI sequencing for 21 individuals from Réunion, the second youngest volcanic island in the region, all Réunion haplotypes fall within the most recently diverged clade (fig. 4).
The third line of evidence is that the diversity observed between populations is strongly correlated with the distance between landmasses. The presence of seven different populations within Ms, strongly related to the island of collection (significant isolation by distance, and TESS structuring) suggests that the radiation occurred as a result of natural processes prior to human arrival in the region. Ancestral expansion of this species from Madagascar and subsequent radiation is supported by gene flow observed between Madagascar and all the other islands, except the Seychelles. Indeed, the levels of gene flow are clearly too low between the Seychelles and the other islands to have homogenised the gene pool of Ms. Isolation by distance appears to limit gene flow, such that Nm is usually less than one.
Despite the fact that our data support an indigenous status for Ms in the SWIO islands, the highest nucleotide diversity observed for Ms populations, for COI, comes from individuals from mainland Africa (Uganda). This suggests that Ms population present in all the islands of the SWIO, might originate from mainland Africa. Up to now, no other sequences from the Ms are available from other African countries, but obviously we suspect that in the future more Ms individuals could be found in the Eastern part of mainland Africa. In previous studies, B. tabaci (Delatte et al., Reference Delatte, Reynaud, Granier, Thornary, Lett, Goldbach and Peterschmitt2005b; Sseruwagi et al., Reference Sseruwagi, Legg, Maruthi, Colvin, Rey and Brown2005: fig. 4), B, Q and Ms have been found to be monophyletic. B was originally believed to have originated in the Mediterranean countries (Guirao et al., Reference Guirao, Beitia and Cenis1997), while population genetic studies have since shown that the B probably originated in the Middle Eastern/North African region (Frohlich et al., Reference Frohlich, Torres-Jerez, Bedford, Markham and Brown1999). Those two species have sympatric regions especially in northern Africa, such as Morocco (Tahiri et al., Reference Tahiri, Sekkat, Bennani, Granier, Delvare and Peterschmitt2006). Delatte et al. (Reference Delatte, Reynaud, Granier, Thornary, Lett, Goldbach and Peterschmitt2005b) showed that for COI, B and Q are genetically closer to each other than the Ms is to either of them, indicating that the most ancient divergence is of Ms from B and Q. The possibility to have field hybrid populations between B and Ms (Delatte et al., Reference Delatte, David, Granier, Lett, Goldbach, Peterschmitt and Reynaud2006), is also suggesting that both may have a recent common ancestor. Up to now, no references are available showing any fit hybrids between B and Q populations. As a result, we can imagine that these three species originate from a common ancestor on the African continent and subsequently diverged through evolutionary time. Furthermore, genetic differentiation among the Ms populations between islands indicate that the species differentiation could have occurred before human arrival, differences in environment (such as host plants and climate) and corresponding selection pressures occurring after island colonization may account for species divergence.
The analysis of a broad sample of whiteflies in the Indian Ocean provides strong evidence that the B is confined to the islands of Réunion and Mauritius. The very low COI nucleotide diversities observed for the B compared to the Ms suggests a shorter period of presence in the SWIO and, therefore, that it may well have an exotic status in this region. Furthermore, our data demonstrate that the B of Mauritius and Réunion originated from the same population and have undergone little divergence (with no significant departure from HWE between islands and very low null allele frequency observed). The high number of migrants observed with microsatellite data may reflect the recent genetic link between both populations and may not result from ongoing gene flow. Since no B or any other species were found on islands other than Mauritius or Réunion, two new exclusive hypotheses are proposed: (i) the colonisation of B was hindered by distance; (ii) B has different ecological constraints than Ms, and it is unable to become established on the other islands. In the case of the first hypothesis, if B reaches one of the other islands, it is likely to be extremely damaging due to the numerous, and not yet emergent, indigenous begomoviruses present on those islands.
Acknowledgements
We would like to thank D. Abeeluck for samples from Mauritius. We thank Frédéric Chiroleu for his help in the adaptation of the kriging function developed for TESS to our data set. This study was funded by the CIRAD and the Conseil Régional de La Réunion.
Appendix 1. Linkage disequilibrium table for Bemisia tabaci Ms and B.

NS, not significant.

Appendix 2. Plot between nucleotide diversity (π) of the partial COI gene of the Bemisia tabaci Ms and ages (million years) of the different islands of the Indian Ocean region.











