


Introduction
Seed shape (SS), defined as seed length (SL), seed width (SW) and seed thickness (ST), is a morphological trait of soybean (Glycine max L.) that is associated with seed weight and also affects soybean yield (Liang et al. Reference Liang, Li, Wang and Fang2005; Hu et al. Reference Hu, Zhang, Kan, Ma, Zhang, Shi, Hong, Zhang and Yu2013). Nelson & Wang (Reference Nelson and Wang1989) reported that SS in soybean has significant variation among different varieties. Liang et al. (Reference Liang, Li, Wang and Fang2005) analysed the inheritance of SS components (SL, SW and ST) via an incomplete diallelic cross of eight varieties with their F1 and F2 populations, with the results showing that SL was controlled mainly by cytoplasmic effects and that SW and ST were determined mainly by maternal effects. Recently, SS has become an important breeding objective because of market and industry requirements (Liang et al. Reference Liang, Li, Wang and Fang2005). For example, soybean varieties with round SS are often used as food-type soybeans, which are liked by traditional soybean-derived food customers (Salas et al. Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006). Seed shapes are complex and polygenic traits (Salas et al. Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006) with moderate heritability (59–79%, estimated by Cober et al. Reference Cober, Voldeng and Fregeau-Reid1997). The results of Cober et al. (Reference Cober, Voldeng and Fregeau-Reid1997) suggested that a soybean with an ideal SS could be effectively selected from earlier generations of crosses. Traditionally, selection for SS in soybean has been ineffective and complicated by significant genotype × environment (GE) interactions. Thus, a reliable method that selects the ideal SS should be developed.
Recently, genetic mapping with molecular markers and marker-assisted selection have been widely used in soybean breeding programmes. Molecular markers have been used to analyse the genetic basis of SS using linkage or association analysis methods. Salas et al. (Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006) detected a total of 19 significant quantitative trait loci (QTL) for SS on ten chromosomes (Chr or linkage groups (LGs)) via three recombinant inbred line (RIL) populations from three crosses: Minsoy × Archer, Minsoy × Noir1 and Noir1 × Archer. One of these 19 QTL (located in simple sequence repeat (SSR) marker Satt578 on Chr4 (LG C1)) could be detected across three populations and two environments, and six were stable in at least two populations in both environments. Hu et al. (Reference Hu, Zhang, Kan, Ma, Zhang, Shi, Hong, Zhang and Yu2013) found that six QTL and seven single nucleotide polymorphisms were associated with SS using a RIL population from a cross between Kefeng1 and Nannong1138–2 and 219 cultivated soybean accessions via combination linkage with association analyses. Niu et al. (Reference Niu, Xu, Liu, Yang, Wei, Xie and Zhang2013) identified 59 main-effect QTL and 31 QTL-by-environment interactions for SS and its components, including SL, SW and ST, through association analyses. Of these identified QTL, only a few have been fine-mapped. Xie et al. (Reference Xie, Niu, Zhang, Bu, Zhang, Geng, Feng and Zhang2014) fine-mapped a QTL (located in the Satt640–Satt422 interval on Chr6) in an RIL population from a cross between Lishuizhongzihuang and Nannong493–1; the results showed that eight candidate genes were found to be associated with SS. Quantitative trait loci/SS-associated genes have been verified and cloned in some crops such as rice GS3 (Fan et al. Reference Fan, Xing, Mao, Lu, Han, Xu, Li and Zhang2006; Mao et al. Reference Mao, Sun, Yao, Wang, Yu, Xu, Li and Zhang2010); GS5 (Li et al. Reference Li, Yan, Agrama, Jia, Shen, Jackson, Moldenehauer, McClung and Wu2011a , Reference Li, Fan, Xing, Jiang, Luo, Sun, Shao, Xu, Li, Xiao, He and Zhang b ); qGW5 (Song et al. Reference Song, Huang, Shi, Zhu and Lin2007); GW8 (Wang et al. Reference Wang, Wu, Yuan, Liu, Liu, Lin, Zeng, Zhu, Dong, Qian, Zhang and Fu2012), tomato ovate (Liu et al. Reference Liu, Van Eck, Cong and Tanksley2002) and sun (van der Knaap & Tanksley Reference Van Der Knaap and Tanksley2001) and Arabidopsis AP2 (Jofuku et al. Reference Jofuku, Omidyar, Gee and Okamuro2005; Ohto et al. Reference Ohto, Fischer, Goldberg, Nakamura and Harada2005); MINI3 (Zhou & Ni Reference Zhou and Ni2010); IKU1 (Wang et al. Reference Wang, Garcia, Zhang, Feng, Chauchury, Berger, Peacock, Dennis and Luo2010); IKU2 (Zhou et al. Reference Zhou, Zhang, Kang, Zhao, Zhang and Ni2009); SHB1 (Sun et al. Reference Sun, Shantharaj, Kang and Ni2010); AFR2 (Schruff et al. Reference Schruff, Spielman, Tiwari, Adams, Fenby and Scott2006). In soybean, SS QTL are seldom verified in other populations or cloned; only a few genes have been proven to affect SW and SL (Singh et al. Reference Singh, Fu, El-Habbak, Navarre, Ghabrial and Kachroo2011). However, to our knowledge, little research has been performed to study the molecular mechanism regulating the SS components of soybean varieties in north-eastern China.
The objective of the present study was to identify QTL associated with SS in the RIL population resulting from the cross Dongnong46 × L-100 in multiple environments using SSR markers.
Materials and methods
Plant materials
Ten F1 plants from Dongnong46 (developed by Northeast Agricultural University, Harbin, China) × L-100 (a semi-wild line in north-eastern China) were self-fertilized to produce 129 F2 lines, respectively. These F2 lines were self-pollinated and each line was advanced up to the F5 and F8 generations by single seed descent. So, this mapping population consisted of 129 F2-derived F5–8 (F2:5−8) RIL derived from a cross between Dongnong46 and L-100. L-100 exhibited lower SL (5·83 mm), SW (3·91 mm) and ST (3·29 mm). Dongnong46 had higher SL (7·62 mm), SW (6·87 mm) and ST (6·01 mm). The mutual ratios of SL, SW and ST, including seed length-to-weight (SLW, calculated as SL/SW), seed length-to-thickness (SLT, calculated as SL/ST) and seed weight-to-thickness (SWT, calculated as SW/ST), were also calculated to evaluate SS. L-100 had higher SLW (1·48), SLT (1·78) and SWT (1·24). Dongnong46 had lower SLW (1·10), SLT (1·27) and SWT (1·14).
Field experiments
Field trials were conducted at Harbin (44·15°N, 130·07°E, fine-mesic Chernozem soil) in 2013, 2014 and 2015, at Hulan (46·04°N, 126·73°E, fine-mesic Chernozem soil) in 2013, 2014 and 2015, and at Acheng (45·33°N, 127·00°E, fine-mesic Chernozem soil) in 2013, 2014 and 2015. Seeds were planted 6 cm apart in a single row that was 3 m long, with 0·65 m between rows; three replications were included using a randomized complete block design. At maturity, 20 plants from each line in each plot, used as seed source, were harvested to evaluate SS components.
Evaluation of phenotypic values
Seed length, SW and ST were measured using digital Vernier callipers according to the methods described by Xie et al. (Reference Xie, Niu, Zhang, Bu, Zhang, Geng, Feng and Zhang2014): SLW, SLT and SWT were calculated as SL/SW, SL/ST and SSW/ST.
Simple sequence repeat analyses
Total DNA from the RIL was isolated from freeze-dried leaf tissue via the Cetyltrimethylammonium bromide (CTAB) method (Han et al. Reference Han, Teng, Yu, Poysa, Anderson, Qiu, Lightfoot and Li2008). A total of 727 SSR markers evenly covering all 20 chromosomes (linkage groups) of soybean were selected in conducting the SSR analysis. The polymerase chain reaction (PCR) reaction was conducted according to Han et al. (Reference Han, Teng, Yu, Poysa, Anderson, Qiu, Lightfoot and Li2008), with a minor modification. It was performed in a volume of 20 µl containing 2 µl 10 × PCR buffer, 1·5 µl magnesium chloride (MgCl2) (25 mm), 0·3 µl deoxyribonucleotide triphosphate (dNTP) mixture (10 mm), 0·2 µl Taq polymerase enzyme (10 units/μl), 2 µl SSR primer (2 µm), 2 µl genomic DNA (50 ng), and 12 µl double-distilled water. The amplification temperature protocol included 2 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 47 °C, 30 s at 72 °C, then 5 min at 72 °C. Polymerase chain reaction products were detected on a 6% denatured polyacrylamide gel using the rapid silver staining method (Han et al. Reference Han, Teng, Yu, Poysa, Anderson, Qiu, Lightfoot and Li2008).
Linkage analysis
Linkage and the genetic distance between SSR markers were calculated via Mapmaker 3·0b (Lander et al. Reference Lander, Green, Abrahamson, Barlow, Daly, Lincoln and Newburg1987). The commands including ‘group’, ‘map’, ‘sequence’, ‘lod table’, ‘try’ and ‘compare’, were used for constructing the linkage groups. The error detection ratio was set at 1%. The Haldane mapping function was used with a minimum logarithm of the odds (LOD) score of 3·0 and a maximum distance of 50 cM. The Kosambi mapping function was used to calculate genetic distances with a minimum LOD score of 3·0 and a maximum distance of 50 cM, and the genetic map were drawn with MapChart (Voorrips Reference Voorrips2002).
Data analysis
The broad-sense heritability of SL, SW, ST, SLW, SLT and SWT was calculated as described by Blum et al. (Reference Blum, Klueva and Nguyen2001). Quantitative trait loci were identified using single-factor analysis of variance (PROC GLM, SAS) as described by Primomo et al. (Reference Primomo, Poysa, Ablett, Jackson, Gijzen and Rajcan2005), based on the SL, SW, ST, SLW, SLT and SWT values of the RIL in each tested environment. The interaction between the QTL and nine different tested environments was analysed using genotype × trait (GT) biplot methodology (Yan Reference Yan2001).
Results
Phenotypic variation
Seed shape components, including SL, SW and ST as well as their mutual ratios SLW, SLT and SWT, were measured and calculated in the RIL population grown across nine different environments (Harbin in 2013, 2014 and 2015, Hulan in 2013, 2014 and 2015 and Acheng in 2013, 2014 and 2015). The genetic parameters of the parents and the RIL population, including mean values, standard deviations, and coefficients of variation, are indicated in Table 1. The SL, SW and ST values of Dongnong46 were significantly (P < 0·05) higher than those of L-100 across the nine environments; however, the SLW, SLT and SWT of Dongnong46 were lower than those of L-100. The ranges of the coefficients of variation for SL, SW and ST, and SLW, SLT and SWT in the RIL population were 0·07–0·12 and 0·07–0·22, which suggested that SS behaved in a relatively stable manner among these nine tested environments (Table 1). Though the SL, SW and ST values of a few RI lines exceeded those of Dongnong46 in the different environments, the SL, SW and ST values of most RI lines were more similar to those of L-100. The transgressive segregation of most RI lines in terms of SLW, SLT and SWT behaved between L-100 and Dongnong46. The heritability of SL, SW and ST in the mapping population was higher (SL: 0·67–0·91, SW: 0·73–0·91 and ST: 0·72–0·90), and SLW, SLT and SWT in the mapping population were relatively moderate (SLW: 0·61–0·76, SLT: 0·58–0·66 and SWT: 0·40–0·51). Shaprio–Wilk tests showed that the frequency distributions of SL, SW, ST, SLW, SLT and SWT in this mapping population were continuous (W = 0·86, not significant (NS); W = 0·83, NS; W = 0·80, NS; W = 0·72, NS; W = 0·83, NS; W = 0·77, NS). Both the skew and kurtosis values of these six SS traits, including SL, SW, ST SLW, SLT and SWT, were <1·0 in most environments, which fit an approximately normal distribution.
Table 1. Range, average, standard deviation (s.d.), coefficient of variation (CV), skewness and kurtosis for seed shape of recombinant inbred lines (RIL) under multiple environments

BSH, broad-sense heritability; SL, seed length; SW, seed width; ST, seed thickness; SLW, seed length-to-width; SLT, seed length-to-thickness; SWT, seed width-to-thickness.
Construction of genetic linkage map
To identify SSR markers associated with SS, more than 700 SSR markers were used to analyse polymorphisms between Dongnong46 and L-100, and a total of 260 polymorphic SSR markers were obtained. These SSR markers were further used to screen the RIL population, and 213 polymorphic SSR markers in the RIL population were found. These 213 SSR markers were distributed on 18 chromosomes (LG) defined by Cregan et al. (Reference Cregan, Jarvik, Bush, Shoemaker, Lark, Kahler, Van Toai, Lohnes, Chung and Specht1999); Song et al. (Reference Song, Marker, Shoemaker, Lark, Concibido, Delannay, Specht and Cregan2004) and Hyten et al. (Reference Hyten, Choi, Song, Specht, Carter, Shoemaker, Hwang, Matukumalli and Cregan2010) and were used to construct a molecular genetic linkage group. The map developed encompassed approximately 3623·39 cM, with an average distance of 17·01 cM between markers (data not shown). The longest and shortest distance in this map was 510·24 cM (Chr.5 (LG A1)) and 31·70 cM (Chr.14 (LG B2)), respectively, which included 47 and three SSR markers, respectively. Chr.5 (LG A1) had the most SSR markers and Chr.14 (LG B2) the least.
Quantitative trait loci associated with seed shape
Five QTL, qSL-1 (Satt150), qSL-2 (Satt353), qSL-3 (Satt052), qSL-4 (13_0102) and qSL-5 (Satt514), associated with SL were located on Chr7 (LG M), Chr12 (LG H), Chr12 (LG H), Chr13 (LG F) and Chr17 (LG D2), respectively (Fig. 1 and Table 2). Among them, qSL-1 explained 2·29, 2·00 and 5·43% of the phenotypic variation at Harbin in 2013, 2014 and 2015, respectively, 6·16 and 1·91% of the phenotypic variation at Hulan in 2013 and 2014, respectively, and 7·66% of the phenotypic variation at Acheng in 2015. qSL-2 explained 4·43% of the observed phenotypic variation at Acheng in 2013, 9·98% of the observed phenotypic variation at Hulan in 2014, and 10·81 and 5·54% of the phenotypic variation at Harbin in 2014 and 2015, respectively. The phenotypic contribution of qSL-3 was 22·16 and 14·11% at Hulan in 2013 and 2014, respectively, 17·64, 15·38 and 12·44% at Acheng in 2013, 2014 and 2015, respectively, and 15·09 and 10·82% at Harbin in 2014 and 2015, respectively. qSL-4 explained 5·66 and 1·74% of the phenotypic variation at Hulan in 2013 and 2014, respectively, 9·00 and 3·59% of the phenotypic variation at Acheng in 2013 and 2014, respectively, and 2·33 and 10·98% of the phenotypic variation at Harbin in 2014 and 2015, respectively. qSL-5 could explain 5·65% of the phenotypic variation at Harbin in 2013, 5·01 and 7·74% of the phenotypic variation at Hulan in 2015 and 2013, respectively, and 5·54% of the phenotypic variation at Acheng in 2014.
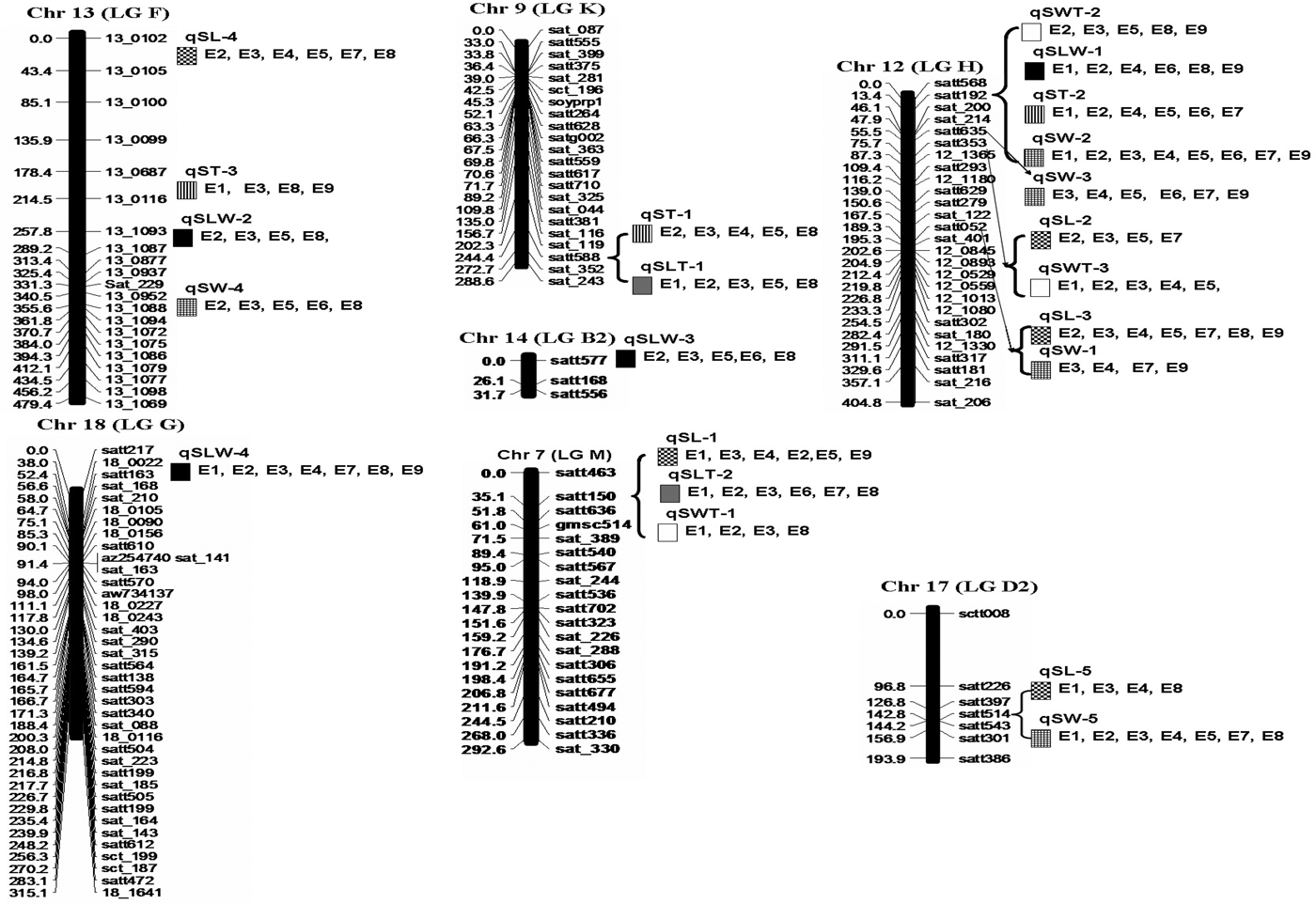
Fig. 1. Genomic locations of the identified quantitative trait loci (QTL) affecting seed shape (SS) components. The map distances in cM are shown on the left. The QTL locations are indicated on the right. ![]() : seed length (SL),
: seed length (SL), ![]() : seed width (SW),
: seed width (SW), ![]() : seed thickness (ST),
: seed thickness (ST), ![]() : seed length-to-width (SLW),
: seed length-to-width (SLW), ![]() : seed length-to-thickness (SLT),
: seed length-to-thickness (SLT), ![]() : seed width-to-thickness (SWT). E1: at Harbin in 2013, E2: at Harbin in 2014, E3: at Harbin in 2015, E4: at Hulan in 2013, E5: at Hulan in 2014, E6: at Hulan in 2015, E7: at Acheng in 2013, E8: at Acheng in 2014, E9: at Acheng in 2015.
: seed width-to-thickness (SWT). E1: at Harbin in 2013, E2: at Harbin in 2014, E3: at Harbin in 2015, E4: at Hulan in 2013, E5: at Hulan in 2014, E6: at Hulan in 2015, E7: at Acheng in 2013, E8: at Acheng in 2014, E9: at Acheng in 2015.
Table 2. Markers associated with seed shape of soybean in multiple environments
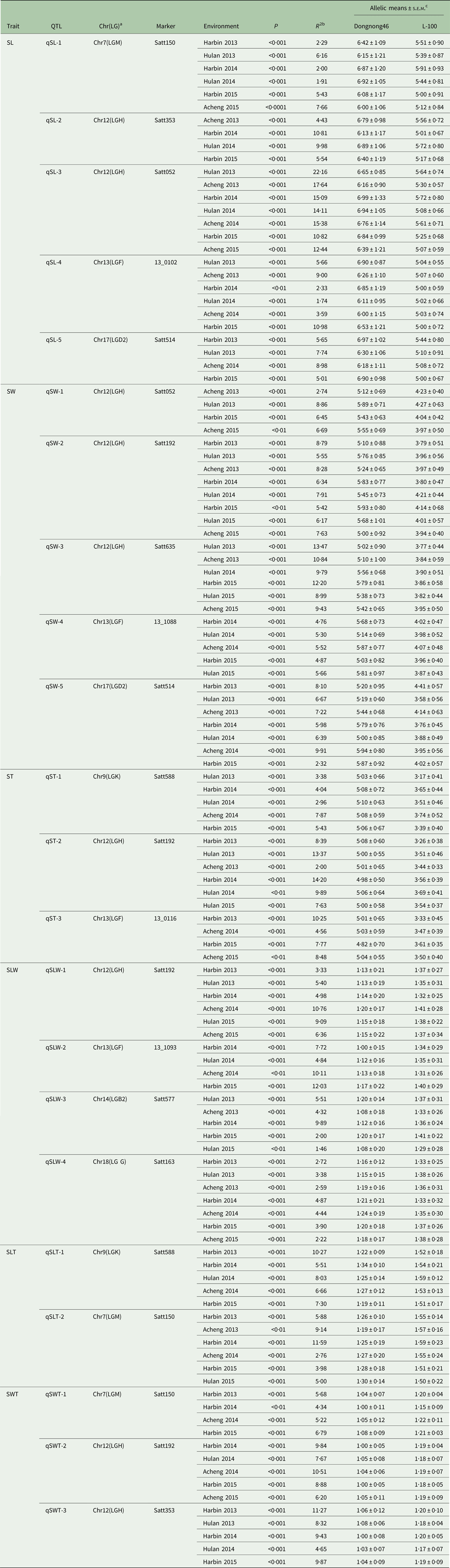
QTL, quantitative trait loci; s.e.m., standard error of the means, SL, seed length; SW, seed width; ST, seed thickness; SLW, seed length-to-width; SLT, seed length-to-thickness; SWT, seed width-to-thickness.
a Chr(LG) indicates the chromosome (linkage group).
b R 2 is R-squared or the proportion of the phenotypic data explained by the marker locus.
c s.e.m. (standard error of the mean): s.d.√N; where N is the number of individuals with each allele.
Five QTL, qSW-1 (Satt052), qSW-2 (Satt192), qSW-3 (Satt635), qSW-4 (13_1088) and qSW-4 (Satt514), were associated with SW and located on Chr12 (LG H), Chr12 (LG H), Chr12 (LG H), Chr13 (LG F) and Chr17 (LGD2), respectively (Fig. 1 and Table 2). Of these, qSW-2 could explain 5·42–8·79% of the observed phenotypic variation across eight tested environments. qSW-5 and qSW-3 explained 2–10 and 9–14% of the phenotypic variation across seven and six tested environments, respectively. qSW-1 and qSW-4 could explain 2–9 and 4–6% of the phenotypic variation at four and five tested environments, respectively.
Three QTL underlying ST were detected and mapped to three chromosomes (Chr9 (LG K), Chr12 (LGH), Chr13 (LG F)) (Fig. 1 and Table 2); these QTL explained 2·96–7·87, 2–14·2 and 4·56–10·25% of the phenotypic variation at three locations in three years. Of these QTL, qST-1 (Satt588) and qST-2 (Satt192) were identified in five and six environments, respectively. However, qST-3 (13_0116) was detected in only four environments.
Four QTL, qSLW-1 (Satt192), qSLW-2 (13_1093), qSLW-3 (Satt577) and qSLW-4 (Satt163) that were associated with SLW were identified on Chr12 (LGH), Chr13 (LGF), Chr14 (LGB2) and Chr18 (LGG), respectively. The phenotypic variation ranged from 1·46 to 12·03% at three locations in 3 years (Fig. 1 and Table 2). Of them, qSLW-1, qSLW-3 and qSLW-4 were identified in six, five and seven environments, respectively; however, qSLW-2 was detected in only four environments.
Two QTL, qSLT-1 (Satt588 on Chr9 (LG K)) and qSLT-2 (Satt150 on Chr7 (LGM)) were identified to be associated with SLT (Fig. 1 and Table 2). Of them, qSLT-2 could explain 5·88, 11·59 and 3·98% of the phenotypic variation at Harbin in 2013, 2014 and 2015, respectively; 9·14 and 2·76% of the phenotypic variation at Acheng in 2013 and 2014, respectively, and 5% of the phenotypic variation at Hulan in 2015. The phenotypic contribution of qSLT-1 was 10·27, 5·51 and 7·30% of the phenotypic variation at Harbin in 2013, 2014 and 2015, respectively, 8·03% of the phenotypic variation at Hulan in 2014 and 6·66% of the phenotypic variation at Acheng in 2014.
Three QTL underlying SWT were identified and mapped to two chromosomes, Chr7 (LGM) and Chr12 (LGH) (Fig. 1 and Table 2); these QTL explained 4·34–11·27% of the phenotypic variation at three locations in 3 years. Of these, qSWT-1 (Satt150 on Chr7 (LGM)), qSWT-2 (Satt192 on Chr12 (LGH)) and qSWT-3 (Satt353 on Chr12 (LG H)) were identified in four, five and five environments, respectively.
Stability evaluation of quantitative trait loci associated with seed shape across the tested environments
In the GT biplot analysis evaluating the stability of the QTL associated with SS across the tested environments, nine QTL (identified in more than six environments) were associated with SS components and explained 70% of the total variation in the standardized data (Fig. 2). When the QTL qSL-3, qSL-4, qSLW-4, qSLW-1 and qST-2 were set as the corner QTL for nine tested environments, seven tested environments (at Harbin in 2014, at Harbin in 2015, at Hulan in 2013, at Hulan in 2014, at Acheng in 2013, at Acheng in 2014 and at Acheng in 2015) fell within the sector in which qSL-3 was the best QTL for these seven tested environments (Fig. 2). qSW-2 and qST-2 were the best QTL for two tested environments (at Harbin in 2013 and at Hulan in 2015). The other QTL were not the best for any tested environments.
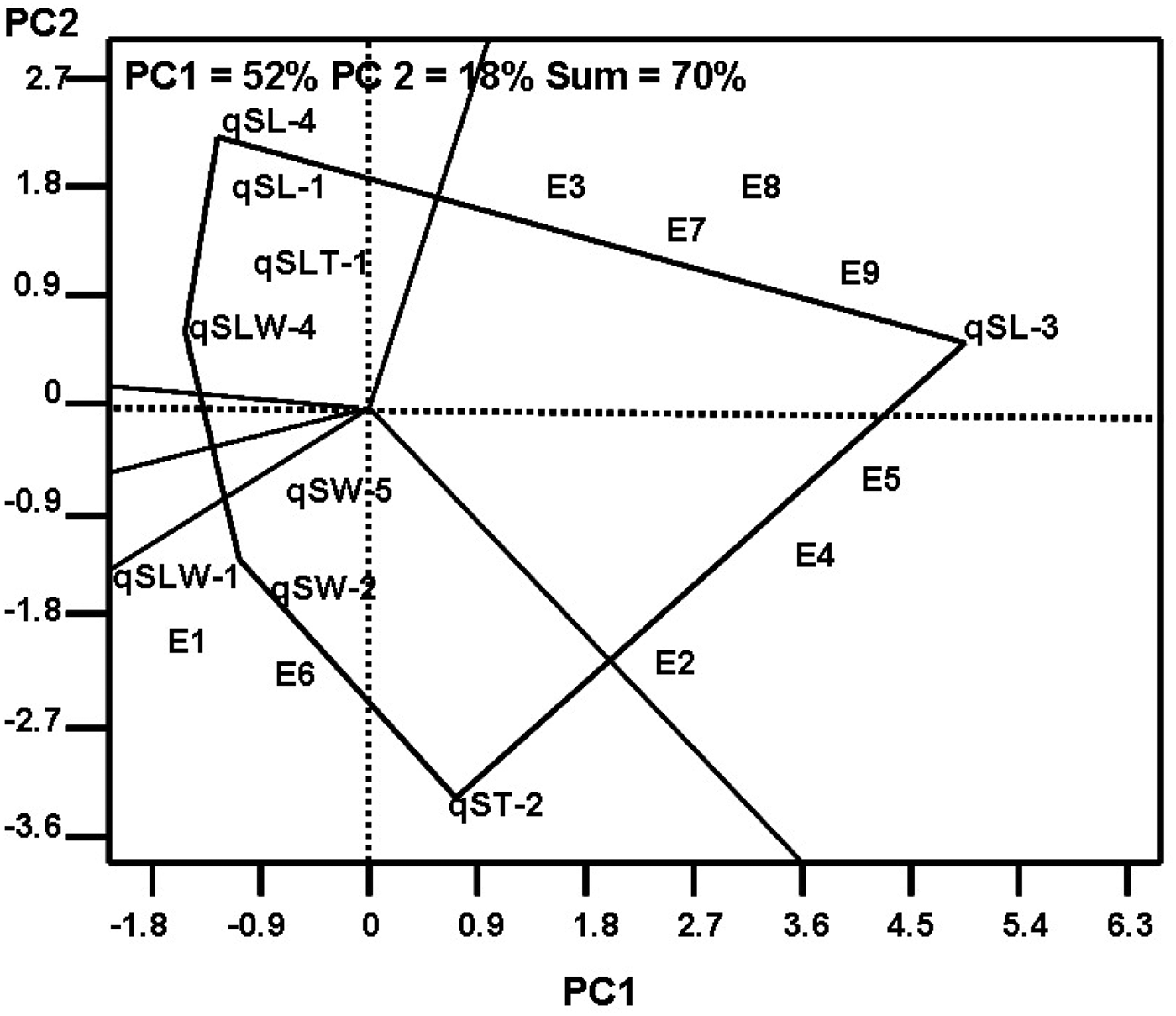
Fig. 2. Genotype × trait (GT) biplot analysis of the relatedness of quantitative trait loci (QTL) and tested environments. PC1: first principle component; PC2: second principle component. E1, at Harbin in 2013; E2, at Harbin in 2014; E3, at Harbin in 2015; E4, at Hulan in 2013; E5, at Hulan in 2014; E6, at Hulan in 2015; E7, at Acheng in 2013; E8, at Acheng in 2014; E9, at Acheng in 2015.
Discussion
Seed shape of soybean, controlled by multiple genes (Salas et al. Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006), could play an important role in determining the weight and appearance of soybeans. Thus, selecting soybean lines with ideal SS is an important breeding target. The results of some studies (Nelson & Wang Reference Nelson and Wang1989; Cober et al. Reference Cober, Voldeng and Fregeau-Reid1997) indicated that SS has a moderate heritability and is relatively stable across environments. The results of the present study also verified those of previous studies (Nelson & Wang Reference Nelson and Wang1989; Cober et al. Reference Cober, Voldeng and Fregeau-Reid1997). Cober et al. (Reference Cober, Voldeng and Fregeau-Reid1997) reported that SS could be selected effectively in early generations. In the present study, transgressive segregation was also found in the RI line. Additionally, the SL, SW, ST, SLW, SLT and SWT values of these transgressive lines were significantly different from those in L-100 and Dongnong46, which were also stable across multiple environments. This phenomenon occurred because these transgressive lines interacted with the positive QTL alleles from parents (Mansur et al. Reference Mansur, Orf, Chase, Jarvik, Cregan and Lark1996; Mian et al. Reference Mian, Bailey, Tamulonis, Shipe, Carter, Parrot, Ashely, Hussey and Boema1996; Orf et al. Reference Orf, Chase, Jarvik, Mausur, Cregan, Adler and Lark1999) or with undetected QTLs or exhibited epistatic interactions. Therefore, it is possible for soybean breeders to select transgressive segregates through molecular markers even if the parents do not have ideal SS. This has been proven for the maturity and yield of soybean through the Minsoy × Noir1 cross by Mansur et al. (Reference Mansur, Orf, Chase, Jarvik, Cregan and Lark1996).
The present study identified five QTL associated with SL, five associated with SW, three with ST, four with SLW, two with SLT, and three with SWT located on four, three, three, four, two and two chromosomes (LG), respectively. The phenotypic variation explained by these QTL ranged from 1·46 to 22·16% for these SS traits in the nine different environments. This result also proved that SS was controlled by multiple genes with minor effects, which was similar to the results of other studies (Salas et al. Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006).
In the present study, qSL-1 (Satt150 on Chr7 (LG M)), which associated with SL across six environments, qSW-5 (Satt514 on Chr17 (LGD2)), which associated with SW across seven environments, and qSWT-1 (Satt150 on Chr7 (LG M)), which associated with SWT across four environments, were identified. These three QTL corresponded to the same interval of three QTL (qSL-7e, R 2 = 7·24%; qSW-17e-1, R 2 = 5·71%; and qSWT-7, R 2 = 10·14%) associated with SL, SW and SWT detected previously by Niu et al. (Reference Niu, Xu, Liu, Yang, Wei, Xie and Zhang2013), who used 257 soybean accessions and three environments in southern China in association analyses. It should be noted that the material tested and identified method reported by Niu et al. (Reference Niu, Xu, Liu, Yang, Wei, Xie and Zhang2013) were different from those in the present study. These three QTL (qSL-1, qSW-5 and qSWT-1) associated with SL, SW and SWT, respectively, were identified in north-eastern China and southern China through linkage and association analysis across mega-environment conditions. This suggests that these three QTL were weakly influenced by genetic background and environment.
In the present study, genetic correlations among these six SS traits were observed, and the same marker was associated with more than one SS trait. For example, Satt192 on Chr12 (LGH) was associated with SW across eight environments, ST across five environments, SLW across six environments and SWT across five environments. It is possible that the different QTL influencing these traits were inherited in clusters as tightly linked loci. This phenomenon was also found in previous studies (Salas et al. Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006; Xu et al. Reference Xu, Li, Li, Wang, Cheng and Zhang2011; Niu et al. Reference Niu, Xu, Liu, Yang, Wei, Xie and Zhang2013). For example, Salas et al. (Reference Salas, Oyarzo-Llaipen, Wang, Chase and Mansur2006) reported that the Satt289–Sat_252 interval simultaneously controlled SL, SW, SL and SWT. However, Aastveit & Aastveit (Reference Aastveit and Aastveit1993) believe that these genetic correlations between common QTL and many traits may be related to the pleiotropy of QTL. Fine mapping was a possible way to answer this issue.
Acknowledgements
The present study was conducted in the Key Laboratory of Soybean Biology of the Chinese Education Ministry, Soybean Research & Development Center (CARS) and the Key Laboratory of Northeastern Soybean Biology and Breeding/Genetics of the Chinese Agriculture Ministry and was financially supported by the National Key R & D Program for Crop Breeding (grant no. 2016YFD0100300), the Heilongjiang Provincial Natural Science Foundation (C2015011), the 948 Project (grant no. 2015-Z53), the Youth Leading Talent Project of the Ministry of Science and Technology in China (grant no. 2015RA228), the Chinese National Natural Science Foundation (grant nos 31471517, 31671717), the ‘Academic Backbone’ Project of Northeast Agricultural University (grant no. 15XG04).




