


Introduction
Social anxiety disorder (SAD) is one of the most prevalent anxiety disorders, often has an early age of onset, a chronic course and is associated with substantial disability and co-morbidity with other emotional disorders and substance abuse (Stein & Stein, Reference Stein and Stein2008).
SAD is widely acknowledged to run in families and possibly have a genetic, or partly genetic cause. Family studies indicate that SAD is 2–9 times more common in first-degree relatives of SAD probands compared to first-degree relatives of healthy controls (Reich & Yates, Reference Reich and Yates1988; Fyer et al. Reference Fyer, Mannuzza, Chapman, Liebowitz and Klein1993; Stein et al. Reference Stein, Chartier, Hazen, Kozak, Tancer, Lander, Furer, Chubaty and Walker1998; Lieb et al. Reference Lieb, Wittchen, Höfler, Fuetsch, Stein and Merikangas2000; Tillfors et al. Reference Tillfors, Furmark, Ekselius and Fredrikson2001; Merikangas et al. Reference Merikangas, Lieb, Wittchen and Avenevoli2003; Coelho et al. Reference Coelho, Cooper and Murray2007). These previous family studies were carefully conducted but also had important limitations. First, families were primarily recruited from specialist anxiety clinics, potentially resulting in the inclusion of more severe and impaired cases. Families with several affected members may have been more likely to participate in such studies, thus inflating the familial risk. Second, although controls were randomly selected using various procedures, families with greater (or fewer) concerns about emotional difficulties may have volunteered to participate more often, leading to inaccurate estimates. These possible biases can optimally be addressed by examining the familial structure of SAD at the population level (Berkson, Reference Berkson1946), recruiting patients from non-specialist clinics and randomly selecting control families from the general population. Third, studies conducted to date were underpowered to calculate risks for relatives with different degrees of genetic relatedness to the proband and different degrees of shared environmental exposures. Consequently, these studies could not disentangle genetic from environmental factors contributing to the observed familiality. Several twin studies of diagnosed SAD cases (Reichborn-Kjennerud et al. Reference Reichborn-Kjennerud, Czajkowski, Torgersen, Neale, Ørstavik, Tambs and Kendler2007) or dimensionally scored SAD symptoms in non-clinical populations (Kendler et al. Reference Kendler, Neale, Kessler, Heath and Eaves1992, Reference Kendler, Karkowski and Prescott1999, Reference Kendler, Myers, Prescott and Neale2001; Warren et al. Reference Warren, Schmitz and Emde1999; Nelson et al. Reference Nelson, Grant, Bucholz, Glowinski, Madden, Reich and Heath2000; Stein et al. Reference Stein, Jang and Livesley2002; Mosing et al. Reference Mosing, Gordon, Medland, Statham, Nelson, Heath, Martin and Wray2009) suggest that both additive genetic (widely ranging between 20% and 55% of the variance) and non-shared environment effects may account for most of the variance in SAD symptoms, with shared environment making a small or negligible contribution, but the wide range of estimates indicates that further research is needed.
SAD substantially overlaps with avoidant personality disorder (AVPD), to the extent that it has been debated whether the latter should be removed from the nomenclature altogether (Stein & Stein, Reference Stein and Stein2008). SAD and AVPD may be part of a severity continuum, with the two disorders being quantitatively, rather than qualitatively, different (Bögels et al. Reference Bögels, Alden, Beidel, Clark, Pine, Stein and Voncken2010). Some family studies have found that SAD and AVPD co-aggregate in the same families (Stein et al. Reference Stein, Chartier, Hazen, Kozak, Tancer, Lander, Furer, Chubaty and Walker1998; Tillfors et al. Reference Tillfors, Furmark, Ekselius and Fredrikson2001; Chavira et al. Reference Chavira, Shipon-Blum, Hitchcock, Cohan and Stein2007) but, given the relatively low prevalence of AVPD, these studies were generally underpowered. A population-based bivariate twin study in Norwegian females found that SAD and AVPD symptoms were influenced by genetic factors common to both disorders, whereas non-shared environmental influences were largely disorder-specific (Reichborn-Kjennerud et al. Reference Reichborn-Kjennerud, Czajkowski, Torgersen, Neale, Ørstavik, Tambs and Kendler2007). Given the paucity of data and limited statistical power of previous investigations, further research is needed to understand the nature of the familial link between SAD and AVPD.
In an attempt to overcome some of these limitations and provide unbiased estimates of family clustering and heritability of SAD and AVPD at the population level, we linked and analyzed data from two Swedish population-based registers and tested four hypotheses: (1) SAD will cluster in families at the population level; (2) the risk of SAD in relatives will increase proportionally to the degree of genetic relatedness to the proband; (3) shared environment effects will be negligible and (4) the risk of AVPD will be higher in relatives of probands with SAD compared to relatives of controls.
Method
Swedish registers
Following approval from the Regional Ethics Committee in Stockholm, we linked two Swedish national registers, using the individual national registration numbers assigned at birth.
The Multi-Generation Register contains information about the identity of biological and adoptive parents of each individual born in Sweden since 1932 (with the mother as informant) or who immigrated to Sweden together with one or both parents before the age of 18 years and lived in Sweden at any time since 1961. Unless the biological/adoptive parents have actually lived in Sweden since 1947, when the national personal identification number was introduced, it is not possible to identify them. The father was defined either as the mother's husband at the time of birth, or the man acknowledged as father by unmarried mothers. With information on parents, it is possible to create family pedigrees for all individuals with relatives at increasing genetic and environmental distances from each index person. For instance, individuals sharing parents may be identified as siblings, and those sharing grandparents as cousins. It is also possible to include non-biological relatives like spouses/partners and adopted children.
The National Patient Register contains diagnostic information about all psychiatric patients treated in Sweden since 1969, with each consultation as a unique record in the register. Initially, it contained information on all inpatient care. From 2001, however, it also includes individuals with outpatient visits to specialist physicians (other than general practitioners) that resulted in one or more psychiatric diagnoses according to the ICD-10. This register has been heavily used for research purposes (Ludvigsson et al. Reference Ludvigsson, Andersson, Ekbom, Feychting, Kim, Reuterwall, Heurgren and Olausson2011).
ICD diagnostic codes
SAD and AVPD probands were defined as individuals identified in the National Patient Register (1997–2009) with at least one diagnosis of social phobia (ICD-10 code F40.1) or AVPD (ICD-10 code F60.6). Note that ICD-10 codes were first introduced in Sweden in 1997 and that previous editions of ICD did not separate different types of phobia.
Main data analyses
The risk of SAD and AVPD in relatives of probands with SAD were compared to the risk in relatives of 10 randomly selected, unaffected control individuals matched by sex, birth year and county of residence at the time of the first recorded SAD diagnosis of the proband. Relatives were also matched by sex and birth year. For instance, for each proband, we detected all possible proband–full-sibling pairs, and randomly selected 10 control–full-sibling pairs matched to proband–sibling pairs by sex and birth year. Because each proband may appear multiple times in different categories (e.g. parent, sibling, cousin) depending on family structure, the matching was done separately for each proband–relative pair to ensure adequate control of cohort/period effects and allow for equal time at risk for proband relatives and control relatives. The matching procedure was used for all available first-, second- and third-degree relatives, as well as non-biological relatives of each proband. First-degree relatives included full siblings, parents, and children. Second-degree relatives included maternal and paternal half-siblings, grandparents and grandchildren, uncles/aunts, and nephews/nieces. Third-degree relatives consisted of first cousins. Non-biological relatives were defined as individuals who had at least one child together (e.g. spouses and partners). There were too few affected twins and adopted cases available for analysis.
Because the data were matched and the outcome dichotomous, we employed a conditional logistic regression model with the proc phreg procedure in SAS, v. 9.3 (SAS/STAT®, 2004). Because several possibly correlated pairs of relatives from every family could be included in the analysis, we adjusted for the non-independence of family members (e.g. several sibling pairs, which share the same parents) by computing corrected (less narrow) confidence intervals (CIs) with a robust sandwich estimator (covsandwich option in phreg) (Lichtenstein et al. Reference Lichtenstein, Yip, Björk, Pawitan, Cannon, Sullivan and Hultman2009; Frisell et al. Reference Frisell, Lichtenstein and Långström2011; Tidemalm et al. Reference Tidemalm, Runeson, Waern, Frisell, Carlström, Lichtenstein and Långström2011; Mataix-Cols et al. Reference Mataix-Cols, Boman, Monzani, Ruck, Serlachius, Langstrom and Lichtenstein2013).
As a complementary measure of familial risk, tetrachoric correlations were calculated from the unmatched data. From this, we estimated additive genetic effects comparing full siblings and maternal half-siblings (assuming that they have similar shared environments but 50% v. 25% genetic similarity, on average). We employed a variation of Falconer's formula (Falconer, Reference Falconer1960):
 $$V_{\rm A} = {\rm 4}(\rho \,{\rm full}\,{\rm siblings} - \rho \,{\rm maternal}\,\,{\rm half} - {\rm siblings}),$$
$$V_{\rm A} = {\rm 4}(\rho \,{\rm full}\,{\rm siblings} - \rho \,{\rm maternal}\,\,{\rm half} - {\rm siblings}),$$
where V A = additive genetic effects and ρ = tetrachoric correlation.
We also examined potential gender effects by separately analysing respective pairs of male–male, male–female, female–female and female–male probands and relatives.
Sensitivity analyses
Although the diagnosis of SAD is usually straightforward, all statistical analyses described above were repeated excluding all individuals with other relevant mental disorders that may potentially lead to differential diagnostic difficulties, namely pervasive developmental disorders (F84), schizophrenia, schizotypal and delusional disorders (F20–F29), paranoid personality disorder (F60.0) and schizoid personality disorder (F60.1).
Results
Sample characteristics
From a total population of 10 667 319 unique individuals who were alive and resident in Sweden during 1997–2009, we identified 18 399 individuals diagnosed with SAD (i.e. ICD-10 social phobia) and 2673 individuals with AVPD. Of these, 679 individuals had been diagnosed with both SAD and AVPD. In cases with a diagnosis of SAD, 3.7% also had a diagnosis of AVPD, whereas in cases of AVPD, 25.4% also had a diagnosis of SAD (Table 1). On average, patients with SAD and AVPD had three contacts with a psychiatrist during the study period [SAD, mean = 3.3 (s.d. = 4.5) contacts; AVPD, mean = 3.1 (s.d. = 4.1) contacts].
Table 1. Characteristics of 18 399 individuals diagnosed with social anxiety disorder (SAD) (ICD-10 social phobia) and 2673 individuals with a diagnosis of avoidant personality disorder (AVPD) identified in the Swedish National Patient Registers between 1997 and 2009

Of the SAD subjects, 88% had at least one lifetime psychiatric co-morbidity; 40.9% had at least one other anxiety disorder [excluding obsessive compulsive disorder (OCD)], 8.3% OCD, 53.2% depression, 26.6% any substance use, 6.4% schizophrenia and 6.7% bipolar disorder. Furthermore, 16.1% had personality disorders other than AVPD. The AVPD probands had even higher rates of psychiatric co-morbidity (94%), particularly anxiety disorders (77.1%), depression (61.5%) and other personality disorders (23.4%).
Familial risk of SAD
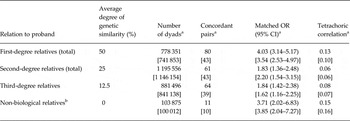
First-degree relatives of individuals with SAD (50% genetic similarity) had a significantly higher risk of having SAD than second-degree relatives (25% genetic similarity) and third-degree relatives (12.5% genetic similarity). In turn, the odds ratios (ORs) for second-degree relatives were significantly higher than for third-degree relatives (Table 2 and Fig. 1). This pattern is strongly suggestive of genetic influences on the liability to SAD.

Fig. 1. Odds ratios [with 95% confidence intervals (CI)] for social anxiety disorder (SAD) among relatives with differing genetic and environmental distances to all diagnosed SAD cases in the Swedish National Patient Register (1997–2009) compared to matched population controls. Each individual in the study population may appear multiple times in different categories (e.g. parent, sibling, cousin) depending on family pedigree. Circles indicate first-degree relatives; diamonds, second-degree relatives; triangles, third-degree relatives; and squares, non-biological relatives. Non-biological relatives are individuals who have at least one child together with the proband with SAD.
Table 2. Risks of social anxiety disorder in relatives of probands diagnosed with social anxiety disorder in Sweden (1997–2009, N = 18 399) compared to relatives of matched controls, and tetrachoric correlations

OR, Odds ratio; CI, confidence interval.
a Individuals who have at least one child together with the proband.
Shared environmental influences on SAD appeared to be considerably less important. Full siblings, parents, and children of SAD probands (all with 50% genetic similarity but siblings assumed to have more shared environment as they grew up together in the same family during about same period of time) had comparable risks. Additionally, the risks for full siblings (50% genetic similarity) were significantly higher than that for maternal half-siblings (25% genetic similarity), despite similar environmental exposure. Furthermore, maternal and paternal half-siblings had comparable risks of having a diagnosis of SAD [both with 25% genetic similarity but with maternal half-siblings sharing more environment as most (90%) children in Sweden continue to live with their mother after parental divorce or separation] (Statistics Sweden, 1994). Finally, first cousins (12.5% genetic similarity) had a 72% higher risk of having SAD compared to controls, despite no or marginal shared environmental exposure with the SAD proband.
Non-biological relatives (i.e. spouses or partners sharing at least one child in common with the probands) were significantly more likely to have SAD themselves, compared to non-biological relatives of matched controls. In fact, their OR (4.01, 95% CI 3.26–4.95) was significantly higher than the risk for biological second- and third-degree relatives.
Tetrachoric correlations and heritability estimates
Tetrachoric correlations were substantially higher for first-, compared to second- and third-degree relatives (Table 2). Contrasting full siblings and maternal half-siblings (assuming similar shared environments but 50% v. 25% genetic similarity, on average), allowed us to estimate the genetic contribution to SAD liability to be approximately 0.56 (or 56%).
Gender effects
Analyses by gender of the proband and gender of the relative showed comparable ORs for male–male, male–female, female–male and female–female dyads (online Supplementary Table S1).
AVPD in relatives of SAD probands
First-degree relatives of individuals with SAD were approximately 3–4 times more likely to have a diagnosis of AVPD. Second- and third-degree relatives had approximately a 2-fold increased risk of AVPD. Finally, mating partners of individuals with SAD were approximately 3–4 times more likely to have a diagnosis of AVPD. These associations largely remained unchanged after the exclusion of probands and relatives with a double diagnosis of SAD and AVPD (Table 3).
Table 3. Risks of avoidant personality disorder (AVPD) in relatives of probands with social anxiety disorder (SAD) compared to relatives of matched controls, before and after exclusion of probands with AVPD and relatives with SAD
OR, Odds ratio; CI, confidence interval.
a The values in square brackets correspond to analyses excluding probands with AVPD and relatives with SAD.
b Individuals who have at least one child together with the proband.
SAD and AVPD in relatives of AVPD-only probands
The risk of SAD in relatives of AVPD-only probands (N = 1994) was approximately the same as the risk of AVPD in relatives of SAD probands, although the statistical power was more limited, resulting in larger CIs (online Supplementary Table S2). Additionally, the risk of AVPD in relatives of AVPD-only probands, was similar to the risk of SAD in relatives of SAD probands, although again this analysis was underpowered (online Supplementary Table S2).
Sensitivity analyses
The main findings reported above did not change substantially after the exclusion individuals who had a lifetime diagnosis of pervasive developmental disorders, schizophrenia, schizotypal and delusional disorders, paranoid personality disorder, or schizoid personality disorder (online Supplementary Table S3).
Discussion
Confirming previous, much smaller, family studies primarily conducted in clinical settings, SAD was significantly more prevalent among biological relatives of SAD probands than in relatives of matched population controls. Further, the risk of SAD in relatives increased significantly with increasing genetic relatedness to the proband. Previous twin studies of categorically diagnosed SAD (Reichborn-Kjennerud et al. Reference Reichborn-Kjennerud, Czajkowski, Torgersen, Neale, Ørstavik, Tambs and Kendler2007) or dimensionally measured SAD symptoms in non-clinical samples (Kendler et al. Reference Kendler, Neale, Kessler, Heath and Eaves1992, Reference Kendler, Karkowski and Prescott1999, 2001; Warren et al. Reference Warren, Schmitz and Emde1999; Nelson et al. Reference Nelson, Grant, Bucholz, Glowinski, Madden, Reich and Heath2000; Stein et al. Reference Stein, Jang and Livesley2002; Mosing et al. Reference Mosing, Gordon, Medland, Statham, Nelson, Heath, Martin and Wray2009) estimated the additive genetic contribution to range between 20% and 55%. Our approximate heritability estimates (56%) are at the higher end of that range.
Together with the previous twin literature (Kendler et al. Reference Kendler, Neale, Kessler, Heath and Eaves1992, Reference Kendler, Karkowski and Prescott1999, 2001; Warren et al. Reference Warren, Schmitz and Emde1999; Nelson et al. Reference Nelson, Grant, Bucholz, Glowinski, Madden, Reich and Heath2000; Stein et al. Reference Stein, Jang and Livesley2002; Reichborn-Kjennerud et al. Reference Reichborn-Kjennerud, Czajkowski, Torgersen, Neale, Ørstavik, Tambs and Kendler2007; Mosing et al. Reference Mosing, Gordon, Medland, Statham, Nelson, Heath, Martin and Wray2009), our data suggest that SAD runs in families primarily due to genetic factors and that shared environmental factors may make a much smaller contribution to the aetiology of the disorder. Instead, unique or non-shared environmental influences may confer increased risk to developing SAD. Even family factors traditionally considered as ‘shared’ environment, such as marital conflict, could technically be classified as ‘non-shared’ (e.g. parenting being experienced as different among siblings) (Plomin, Reference Plomin2011). The identification of genetic differences in susceptibility to particular environments (gene–environment interactions) in SAD will be an important challenge for the future. Finally, the possibility of gene–environment correlations should also be investigated, as it is plausible that genetic factors could influence the specific environmental experiences of children vulnerable to developing SAD (Jaffee & Price, Reference Jaffee and Price2007).
No gender differences in familial patterns
While SAD tends to be more prevalent in women, and male and female patients with SAD differ on several clinical variables, such as clinical symptoms, co-morbidity patterns and help-seeking behaviours (Weinstock, Reference Weinstock1999; Xu et al. Reference Xu, Schneier, Heimberg, Princisvalle, Liebowitz, Wang and Blanco2012), our results suggest that the familial risk for SAD is comparable in male and female probands, regardless of the sex of the relative. Previous family (Tillfors et al. Reference Tillfors, Furmark, Ekselius and Fredrikson2001) and twin (Kendler et al. Reference Kendler, Jacobson, Myers and Prescott2002) studies of SAD had provided mixed results in this regard but were probably underpowered to examine gender differences. The prediction for molecular genetic research would be that when specific genes associated with SAD are identified, they will be associated with SAD in both sexes and that they will have similar effect sizes in males and females. However, the results do not preclude the role of gender-specific precipitating or maintaining factors, such as those linked to the reproductive cycle in females (Weinstock, Reference Weinstock1999).
Are SAD and AVPD aetiologically related?
Supporting previous, much smaller studies (Stein et al. Reference Stein, Chartier, Hazen, Kozak, Tancer, Lander, Furer, Chubaty and Walker1998; Tillfors et al. Reference Tillfors, Furmark, Ekselius and Fredrikson2001), we found that biological relatives of individuals with SAD were significantly more likely to have a diagnosis of AVPD, even after excluding probands with AVPD and relatives with SAD. The same was true when we examined the risks of SAD in relatives of probands with AVPD. The ORs tended to be significantly higher for first- compared to second- and third-degree relatives, although some of the CIs overlapped. Due to the relatively low prevalence of AVPD, these analyses may have been underpowered to fully separate the various types of relatives, particularly after the exclusion of individuals who had both diagnoses. Thus, we cannot confidently confirm whether the familial link between SAD and AVPD is explained by shared genetic or shared environmental factors. However, combined with previous twin research (Reichborn-Kjennerud et al. Reference Reichborn-Kjennerud, Czajkowski, Torgersen, Neale, Ørstavik, Tambs and Kendler2007), the results suggest that SAD and AVPD are probably phenotypic variants of the same underlying vulnerability. An alternative explanation is that clinicians do not reliably or consistently make the distinction between SAD and AVPD, or regard the latter as a severe variant of the former. In both instances, the implication for molecular genetic research would be that when susceptibility genes are identified for one condition, it would be reasonable to suspect that they may also be involved in the other disorder and vice versa. It is, however, unlikely that these genes will directly underlie these ‘man-made’ diagnoses; rather, these genes are more likely to underlie more basic temperamental dimensions, such as neuroticism or behavioural inhibition, which in turn increase the risk for SAD and AVPD, alongside other emotional disorders (Stein et al. Reference Stein, Chartier, Hazen, Kozak, Tancer, Lander, Furer, Chubaty and Walker1998; Stein & Stein, Reference Stein and Stein2008).
SAD/AVPD in non-biological relatives
Although not hypothesized, non-biological relatives of probands with SAD (defined as individuals who have at least one child together with the proband) were approximately four times more likely to also have a diagnosis of SAD, compared to non-biological relatives of controls. This was also true for AVPD, even after the exclusion of probands with SAD and relatives with AVPD.
To our knowledge, an increased risk of clinically diagnosed SAD or AVPD in spouses/partners of SAD probands has not been reported before. We have recently reported a similar, albeit smaller effect in OCD (Mataix-Cols et al. Reference Mataix-Cols, Boman, Monzani, Ruck, Serlachius, Langstrom and Lichtenstein2013) and other studies have also inconsistently found marital concordance for various psychiatric disorders (Merikangas et al. Reference Merikangas, Prusoff and Weissman1988; Maes et al. Reference Maes, Neale, Kendler, Hewitt, Silberg, Foley, Meyer, Rutter, Simonoff, Pickles and Eaves1998; Dierker et al. Reference Dierker, Merikangas and Szatmari1999). The current design did not enable delineating possible marital interaction effects from assortative mating, or social homogamy. Spouses/partners could potentially become more similar the longer they cohabit. With time, lack of social exposure may result in the originally unaffected spouse/partner becoming more socially anxious and eventually seeking help and receiving a diagnosis. However, this interpretation seems particularly far-fetched, given that the origins of both SAD and AVPD can often be traced back to childhood/adolescence and that onset after age 25 years is very rare (Chavira & Stein, Reference Chavira and Stein2005). Therefore, it is reasonable to assume that majority of affected couples met and had children after the onset of their disorder. Thus, it may be more plausible to attribute the findings to assortative mating; individuals with SAD or AVPD may seek partners with similar characteristics and eventually have children with them. This intriguing possibility and, particularly the health and social consequences for their offspring, should be studied further.
Strengths and limitations
One strength of the present study is the very large population-based sample of SAD and AVPD cases diagnosed in Sweden over more than a decade, all their relatives, as well as carefully matched, randomly selected controls. This ensured minimal risk of selection, recall, and report biases for both SAD and control families. Further, this is the first study to examine the familial risk of SAD across relatives at varying genetic and environmental distances from the probands and to have sufficient power to examine the familial risk of AVPD in relatives of SAD probands after the exclusion of individuals endorsing both diagnoses.
Nevertheless, registers also have limitations. First, individuals diagnosed with SAD in the National Patient Register represent a fraction of all SAD cases in the Swedish population. Many individuals with SAD do not seek help and, thus, may never be diagnosed or treated (Stein & Stein, Reference Stein and Stein2008). Furthermore, the National Patient Register only includes patients seen by specialist physicians; those diagnosed in primary care by general practitioners or other professionals (e.g. psychologists) are not included. Finally, psychiatric outpatients were only included in the register from 2001. Thus, the register may only include the more severe and complex forms of SAD (in our cohort, over 80% of patients had at least one lifetime psychiatric co-morbidity) and our results may not generalize to milder forms of the disorder. Some of these limitations may also apply to previous family studies conducted in specialist centres. The impact of these factors on the current estimates is unknown but the incomplete coverage of SAD cases in the Register should be constant across the families of probands and the families of comparison subjects. It is theoretically possible that having a relative with SAD/AVPD increases the chance of seeking help/receiving a diagnosis, although our findings suggest small or negligible shared environmental effects, which would argue against this possibility. Reassuringly, the familial risks and heritability estimates obtained in our study are not dissimilar to those obtained in previous (traditional) family/twin studies.
A second limitation of our study is that the relevant ICD-10 codes have not been formally validated in the Swedish registers. However, the clinical diagnosis of SAD is often straightforward and the exclusion of individuals with the most obvious deferential diagnoses from our analyses did not affect the results. Third, longitudinal registers are subject to ‘left truncation’ or missing data before the date the register started (social phobia was introduced as a separate disorder in ICD-10, and ICD-10 was only included in Sweden in 1997). However, since we matched for birth year and time at risk, such losses would be similar for both case and control dyads and not affect family risks. Fourth, despite the very large sample size, some of the analyses relating to AVPD were underpowered to separate genetic from shared environmental effects. Fifth, our study could not distinguish between potential subtypes of SAD, although we suspect that most cases included in the register are of the ‘generalized’ type. There is some evidence that, while the generalized subtype of SAD is strongly familial, performance-related social anxiety may not be (Stein et al. Reference Stein, Chartier, Hazen, Kozak, Tancer, Lander, Furer, Chubaty and Walker1998). Non-shared environmental factors, such as being teased at school may play a particularly important role in performance-related social anxiety (Bögels et al. Reference Bögels, Alden, Beidel, Clark, Pine, Stein and Voncken2010). Sixth, despite employing a very large population-based sample of patients and relatives at various genetic and environmental distances from the probands, we still lacked statistical power to model the proportion of variance due to genetic and environmental factors, particularly relating to the association between SAD and AVPD. Finally, although we found strong evidence of marital concordance we could not distinguish between marital interaction, social homogamy and phenotypic assortment effects. Assortative mating could potentially bias the reported heritability estimates for SAD but it does not lead to different interpretations of the familial risks in this study.
Conclusions
With these caveats in mind, our results suggest that SAD is a familial disorder and the observed pattern of familiality is consistent with a likely genetic aetiology. Non-shared environmental influences, rather than those shared within families, are also probably important. AVPD is probably aetiologically related to SAD and may represent a severe variant of the same condition. The finding of possible ‘assortative mating’ in SAD and AVPD is novel and its implications should be explored further.
Supplementary material
For supplementary material accompanying this paper visit http://dx.doi.org/10.1017/S0033291714002116.
Acknowledgements
The Swedish Council for Working Life and Social Research, and the Swedish Research Council supported the study. The funding organizations had no influence on the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Declaration of Interest
None.






