Introduction
Termites are wood-feeding eusocial insects related to cockroaches; they are ecologically important due to their role in the decomposition of organic matter (Bignell & Eggleton, Reference Bignell, Eggleton, Abe, Bignell and Higashi2000). Two termite genera, Reticulitermes and Kalotermes, are distributed in Western Europe. The former is a genus of subterranean termites (Rhinotermitidae) occurring along the Mediterranean and Atlantic coasts, as well as in urban areas, with colonies often composed by diffuse nests and multiple feeding sites connected by underground tunnels (Vargo & Husseneder, Reference Vargo and Husseneder2009). Dry-wood termites of the genus Kalotermes (Kalotermitidae) are more restricted to Mediterranean coasts where they form small colonies in deadwood of various tree species.
Until recently, only one species of Kalotermes was thought to be distributed across Europe, the yellow-necked K. flavicollis. Early studies, though, had already noticed morphometric and physiological variations between Italian, Sardinian and French populations (Luscher, Reference Luscher1956; Springhetti, Reference Springhetti1967). More recently, molecular studies showed that the taxon K. flavicollis is, in fact, composed by at least three main lineages that could represent distinct taxa (Luchetti et al., Reference Luchetti, Bergamaschi, Marini and Mantovani2004, Reference Luchetti, Dedeine, Velonà and Mantovani2013a; Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011). Lineage A includes all samples collected from the Aegean islands (Crete and the Cyclades) to the Italian peninsula. This genetically homogenous lineage was previously termed K. flavicollis sensu stricto (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011; Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a). Lineage SC includes colonies collected in Sardinia and Corsica, while lineage SF comprises those collected in southern France; this third lineage appeared significantly diverging from both lineages A and SC (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011). Furthermore, a fourth, highly divergent lineage was found in sympatry with lineage A in an Italian population and termed lineage B. Interestingly, several colonies were found harbouring mitochondrial DNA haplotypes of both lineages A and B and data on nuclear DNA markers suggested the possibility of interbreeding (Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a).
Beside K. flavicollis, two new Kalotermes species were recently described (Ghesini & Marini, Reference Ghesini and Marini2013, Reference Ghesini and Marini2015). The first new species, Kalotermes italicus, is recognizable by a black (or dark brown) pronotum; it is found in central Italy on both sides of the peninsula (Ghesini & Marini, Reference Ghesini and Marini2013). Interestingly, Becker (Reference Becker1955) described a K. flavicollis form with black pronotum, designated as ‘var. fuscicollis’, and demonstrated that the two color variants can interbreed giving offspring with dark or dark-yellow pronotum. The second new species, Kalotermes phoenicae, was found in Cyprus and along Lebanon and Israel coasts (Ghesini & Marini, Reference Ghesini and Marini2015). Altogether, these studies shed new light on the biodiversity of European Kalotermes termites.
The taxonomy and the distribution pattern of Kalotermes taxa are far from being complete and many issues remain unresolved. For instance, K. italicus was found only in three localities in Central Italy (Ghesini & Marini, Reference Ghesini and Marini2013), although its geographical distribution is probably more extensive. The same is true for the French lineage and nothing is known on the taxonomic and phylogenetic status of Kalotermes from the Iberia peninsula (Maistrello et al., Reference Maistrello, Ocete and López2010). To increase the knowledge about Kalotermes diversity, taxonomy, and distribution in Europe, we sequenced 911 bp of the highly informative cox1/trnL/cox2 mitochondrial DNA region for 43 colonies collected from 28 locations from Spain to southern Italy, including previously unsampled areas of Sicily and Sardinia. Data were then integrated with those provided from previous studies to get a more global picture.
Materials and methods
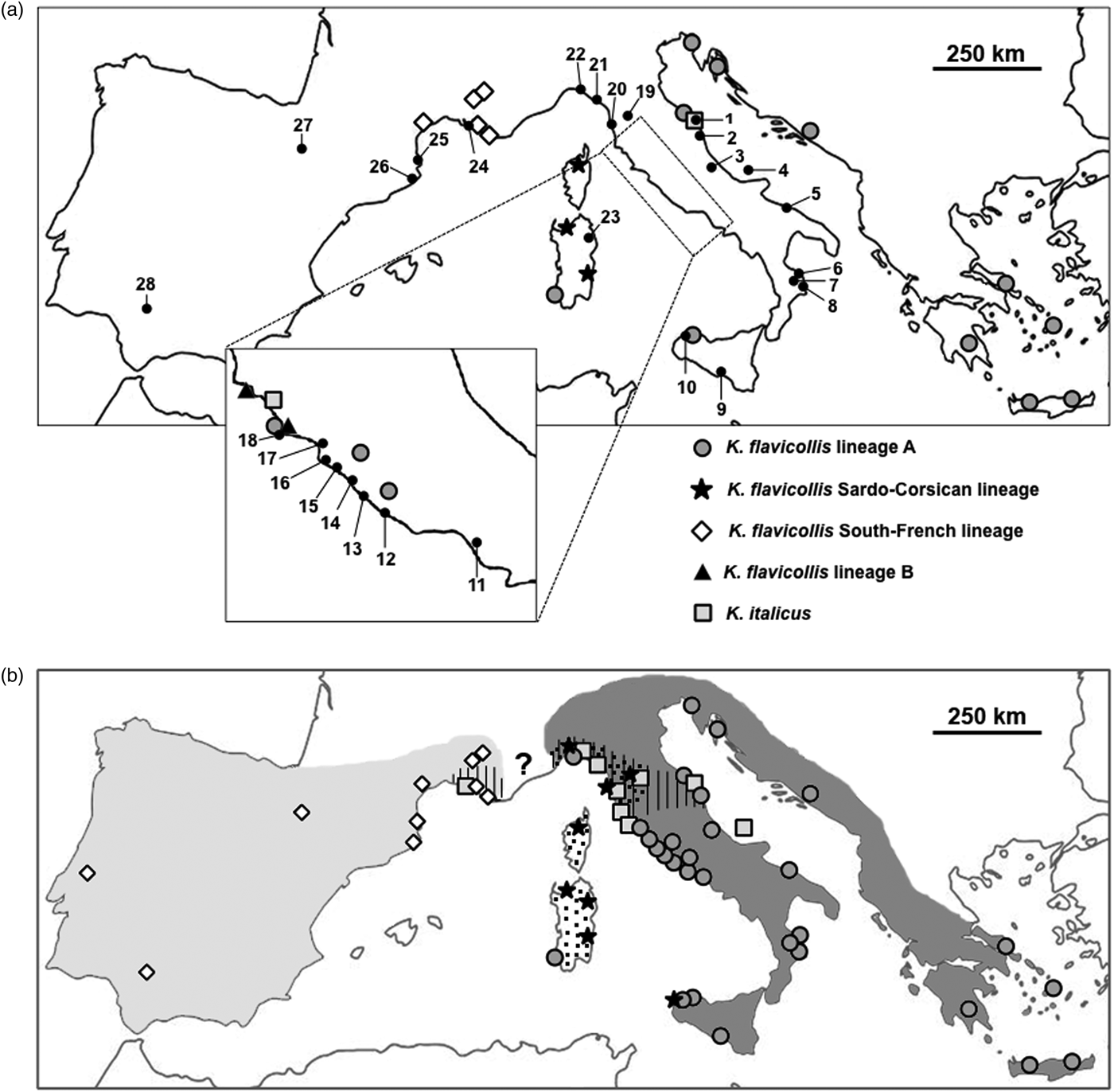
New collection points were chosen to cover previously unsampled or poorly sampled areas. Termites were collected in the field from logs or other pieces of dead wood; most of the specimens were pseudergates (i.e., false workers), which constitute the majority of the colony. For each collection point, pseudergates were carefully taken from the same tunnel and were considered to belong to the same colony. The only exception was the sample of Renzetto (REZ), where we caught swarming alates instead of pseudergates from tunnels. Therefore, we cannot exclude that REZ individuals belong to distinct colonies. All samples were conserved in 100% ethanol until molecular analyses. In total, 43 colonies from 28 localities were analyzed (table 1 and fig. 1a).
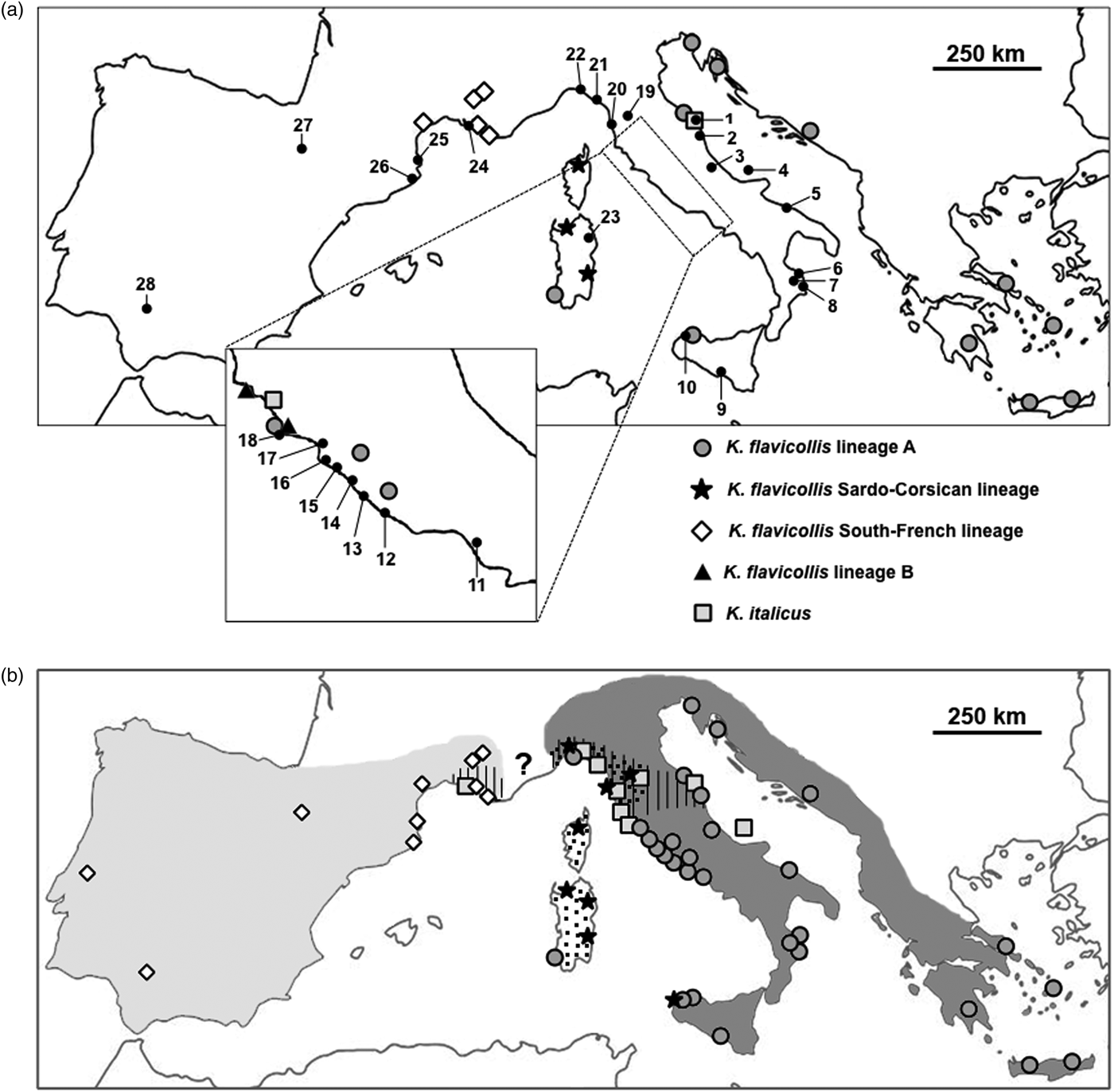
Fig. 1. (a) Kalotermes sampling locations and lineages distribution known so far. Numbers refer to table 1. (b) Summary of European Kalotermes taxa distribution as derived from the present analysis. Light gray area: K alotermes flavicollis lineage IF; dark gray area: K. flavicollis sensu strictu; dotted area: K. flavicollis lineage SC; hatched area: K. italicus. The question mark indicates the lack of information about the distribution boundaries of Kalotermes taxa in that range.
Table 1. List of colony sampling, with scored haplotypes per colony.

1 Only swarming individuals.
2 A single individual sequenced.
Total DNA was isolated using the CTAB method (Doyle & Doyle, Reference Doyle and Doyle1987) from two pseudergates per colony, with the exception of five colonies in which a single individual was analyzed (table 1). A 911 bp mitochondrial fragment encompassing a part of the cox1, the entire length of trnL, and a part of the cox2 regions was PCR amplified and sequenced using the primers C1-J-2797 (5′-CCT CGA CGT TAT TCA GAT TAC C-3′) and TK-N-3785 (5′-GTT TAA GAG ACC AGT ACT TG-3′). Amplification reactions were performed in 50 µl mixtures, using 20 ng of template DNA, with GoTaq DNA polymerase kit (Promega, Madison, WI, USA) following the manufacturer's protocol. The PCR amplification program includes: initial denaturation for 5 min at 95°C; 30 cycles of 30 s at 95°C, 30 s at 50°C, 30 s at 72°C; final extension for 7 min at 72°C. Sanger sequencing of both strands was performed at Macrogen Europe (The Netherlands). Sequences were submitted to Genbank, under accession numbers MF589135–MF589164.
The 81 sequences obtained in this study were analyzed together with sequences taken from previous studies (Luchetti et al., Reference Luchetti, Bergamaschi, Marini and Mantovani2004, Reference Luchetti, Dedeine, Velonà and Mantovani2013a; Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011; Ghesini & Marini, Reference Ghesini and Marini2013), the cox2 sequence from a Portuguese sample of K. flavicollis (GenBank accession number DQ442147; Inward et al., Reference Inward, Vogler and Eggleton2007) and two cox1/trnL/cox2 haplotypes of K. italicus from Grosseto and Portonovo samples (Ghesini & Marini, Reference Ghesini and Marini2013). Moreover, two cox2 sequences belonging to the two divergent lineages of K. phoenicae were added (samples Benouaiti and Kaplica, accession numbers KC914299 and KC914300; Ghesini & Marini, Reference Ghesini and Marini2015). Finally, the cox2 of the New Zealand species K alotermes brouni (accession number AF189104; Thompson et al., Reference Thompson, Miller, Lenz and Crozier2000) was used as outgroup.
Sequence alignment (with Clustal W algorithm), molecular divergence (uncorrected p-distance), and the best substitution model were calculated using MEGA v. 7 (Kumar et al., Reference Kumar, Stecher and Tamura2016). The best substitution model was obtained for each gene individually (cox1: T92; trnL: JC; cox2: HKY + G) and for the entire region (HKY + G + I). Maximum Likelihood phylogenetic tree was calculated using MEGA v. 7, with nodal support based on 100 bootstrap replicates. As MEGA v. 7 does not allow to treat partitions separately, the substitution model HKY + G + I was used for the entire sequence. Bayesian Inference was calculated with MrBayes v. 3.2 (Ronquist et al., Reference Ronquist, Teslenko, van der Mark, Ayres, Darling, Hohna, Larget, Liu, Suchard and Huelsenbeck2012) on a gene-partitioned data set, running for 106 generations and sampling trees every 500 generations. Convergence was reached when the average divergence of split frequencies fell below 0.01. Maximum Likelihood and Bayesian Inference methods yielded a substantially identical topology and similar confidence levels; the Maximum Likelihood tree was therefore used for further analysis.
Haplotype (h D) and nucleotide diversity (π), and Tajima's D analyses were computed with DnaSP v. 5.1 (Librado & Rozas, Reference Librado and Rozas2009). Species delimitation was estimated by using three different methods: single threshold GMYC (Generalized Mixed Yule Coalescent; Fujisawa & Barraclough, Reference Fujisawa and Barraclough2013), PTP (Poisson Tree Processes; Zhang et al., Reference Zhang, Kapli, Pavlidis and Stamatakis2013), and statistical parsimony network (Hart & Sunday, Reference Hart and Sunday2007). As GMYC results appeared to be strictly dependent on the method used for ultrametric tree calculation, we followed Tang et al.’s (Reference Tang, Humphreys, Fontaneto and Barraclough2014) advice and used BEAST v. 1.8 (Drummond & Rambaut, Reference Drummond and Rambaut2007). Moreover, possible biases due to the molecular clock algorithm used (Monaghan et al., Reference Monaghan, Wild, Elliot, Fujisawa, Balke, Inward, Lees, Ranaivosolo, Eggleton, Barraclough and Vogler2009) were overcome with the use of four ultrametric trees obtained with different settings: we built trees using both strict and lognormal relaxed clocks, each implementing either the Yule or the coalescent (with constant population size) tree priors. Calibration was arbitrarily set, imposing the age of the ingroup node to 1.0 and modelling a normal prior distribution with 0.1 of standard deviation; this was done to facilitate the convergence of runs. Each tree was, then, calculated after two runs set at 20 × 106 generations each, sampling every 1000, and the convergence was assessed by estimated sample size >200. The PTP analysis was performed on the web server http://species.h-its.org/, using 5 × 105 Markov chain Monte Carlo generations, burnin = 0.25 and removing the outgroup. Finally, the parsimony network was obtained through TCS v. 1.21 (Clement et al., Reference Clement, Posada and Crandall2000), calculating the 95% connection limit between possible sub-networks: putative specific entities are discriminated based on the number of sub-networks.
Results
Thirty haplotypes, differing from 1 to 65 nucleotide substitutions, were identified (H1-H30; table 1) in the 81 sequences obtained in this study. The most common haplotype (H3) is distributed from Sicily (AGR) up to the Feniglia Reserve (FENc) (table 1).
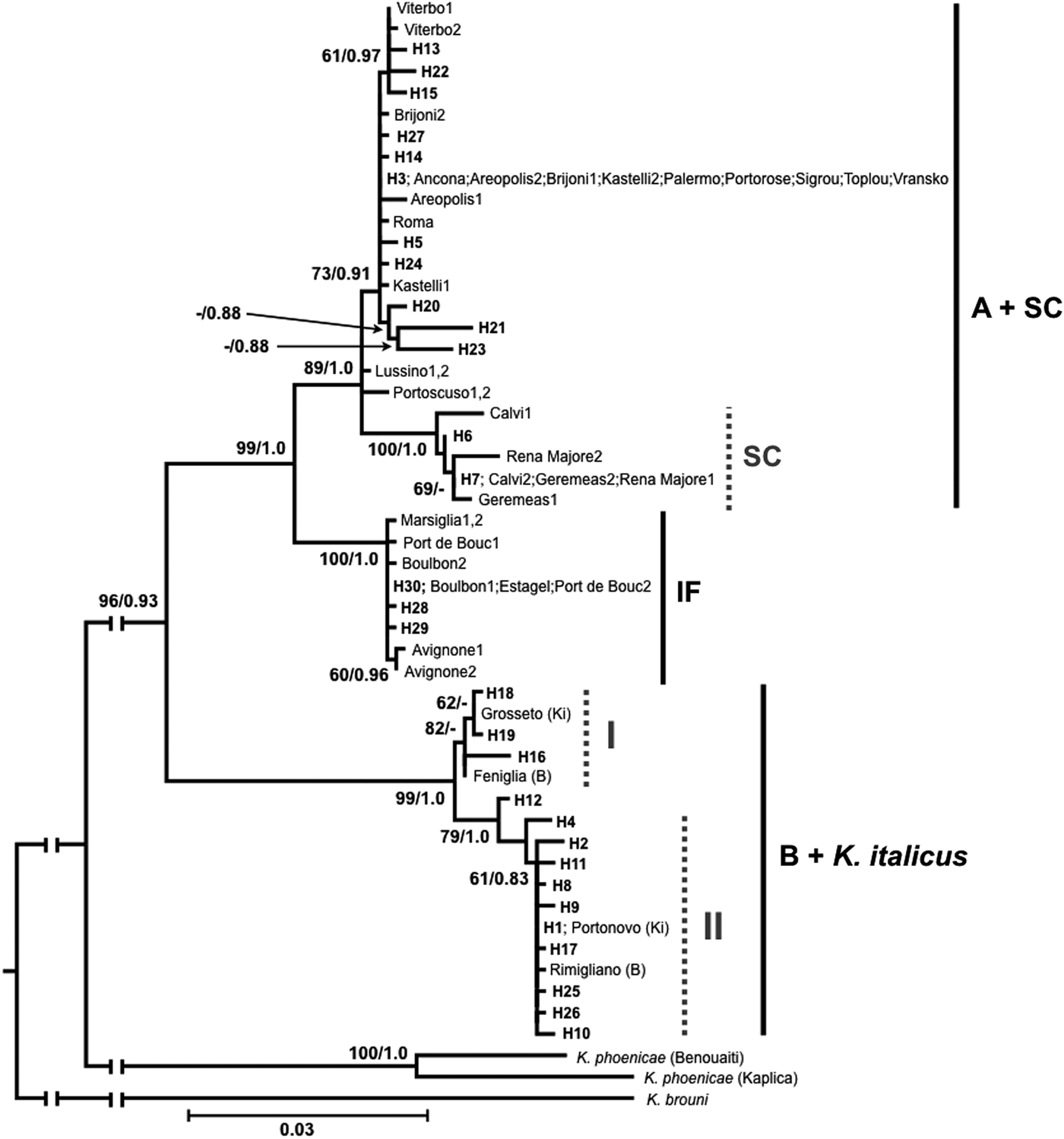
Maximum Likelihood and Bayesian Inference trees were built on haplotypes from all data available (present data; Luchetti et al., Reference Luchetti, Bergamaschi, Marini and Mantovani2004, Reference Luchetti, Dedeine, Velonà and Mantovani2013a; Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011; Ghesini & Marini, Reference Ghesini and Marini2015). Obtained trees gave overlapping topologies and split haplotypes in two main clusters, each further structured into well-supported sub-clusters. These clusters mirror known K. flavicollis lineages and K. italicus species (fig. 2).
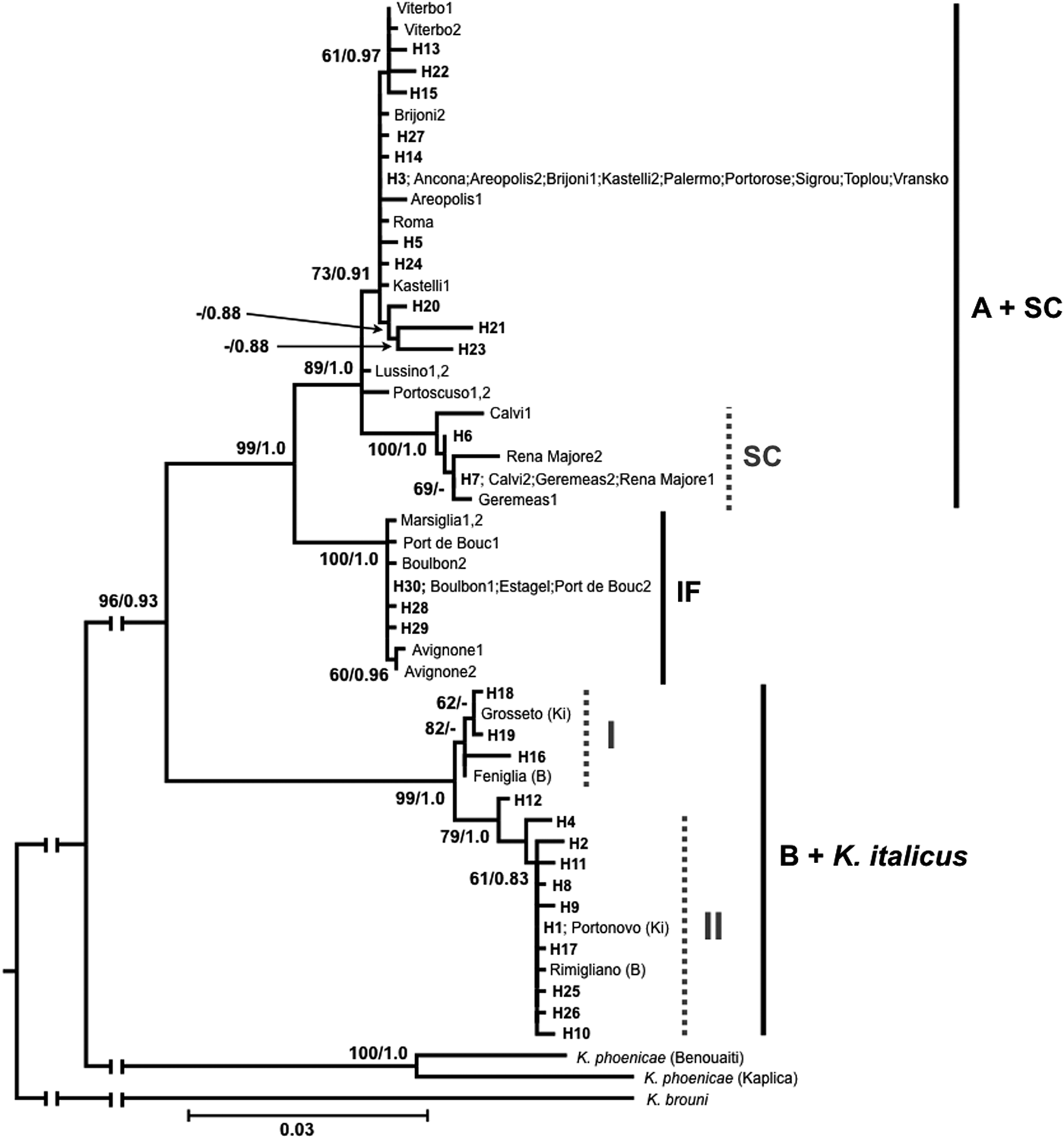
Fig. 2. Maximum Likelihood tree (−lnL = 2953.453) obtained from cox1/trnL/cox2 haplotypes. Bayesian Inference analysis (−lnL = 2973.756) resulted in an overlapping topology. Haplotype codes as in table 1; previously identified haplotypes are reported with the name of the sampling location (consistently with Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011). Lineages are indicated with vertical bars. In the B + K. italicus cluster, samples previously ascribed to lineage B are indicated with ‘B’ in brackets, while those described as Kalotermes italicus are indicated with ‘Ki’. Numbers at nodes are bootstrap values >60%/Bayesian posterior probabilities >0.8. Abbreviations: A, lineage A; B, lineage B; SC, Sardo-Corsican lineage; IF, Ibero-French lineage.
The first main cluster is subdivided into two sub-clusters (fig. 2). The first one embodies haplotypes H3, H5, H13-15, H20-24, H27 and the samples known to belong to K. flavicollis lineage A. It also includes a further small cluster grouping haplotypes H6 and H7 together with the sequences of lineage SC. The second sub-cluster shows a sister relationship with the other one, and groups haplotypes H28-30 together with those of K. flavicollis lineage SF. The cox2 of the Portuguese sample of K. flavicollis is also included in this sub-cluster, being identical to haplotype H30. Given the absence of sub-structures in this lineage, it will be henceforth referred to as the Ibero-French lineage (lineage IF). The second main cluster is also structured in two sub-clusters (fig. 2). The first one (I) includes haplotypes H16, H18, and H19, K. flavicollis lineage B from Feniglia, and K. italicus from Grosseto. The second sub-cluster (II) groups the remaining 10 haplotypes and the other two sequences of K. flavicollis lineage B (Rimigliano) and K. italicus (Portonovo). The two K. phoenicae samples form a single clade that has a sister relationship with the two main clusters (fig. 2).
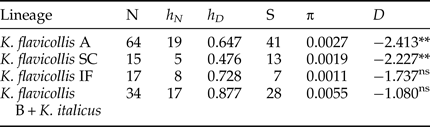
The sequence divergence between clusters and sub-clusters varies widely, ranging from 1.2 to 6.1–6.7% (table S1). K. flavicollis lineage B + K. italicus cluster appeared the most variable based on both haplotype and nucleotide diversity (table 2). The K. flavicollis lineage IF showed a slightly higher haplotype diversity than K. flavicollis lineages A and SC, the latter appearing as the less variable one (table 2). Tajima's D values resulted negative for the four lineages, with only K. flavicollis lineages A and SC showing significant departures from 0 (table 2).
Table 2. Genetic diversity and Tajima's D test for scored Kalotermes lineages.
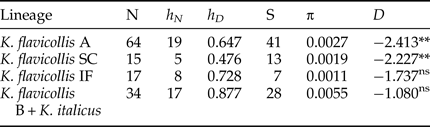
N, number of sequences; h N, number of haplotypes; h D, haplotype diversity; S, number of segregating sites; π, nucleotide diversity; ns, not significant; *P < 0.05; **P < 0.01.
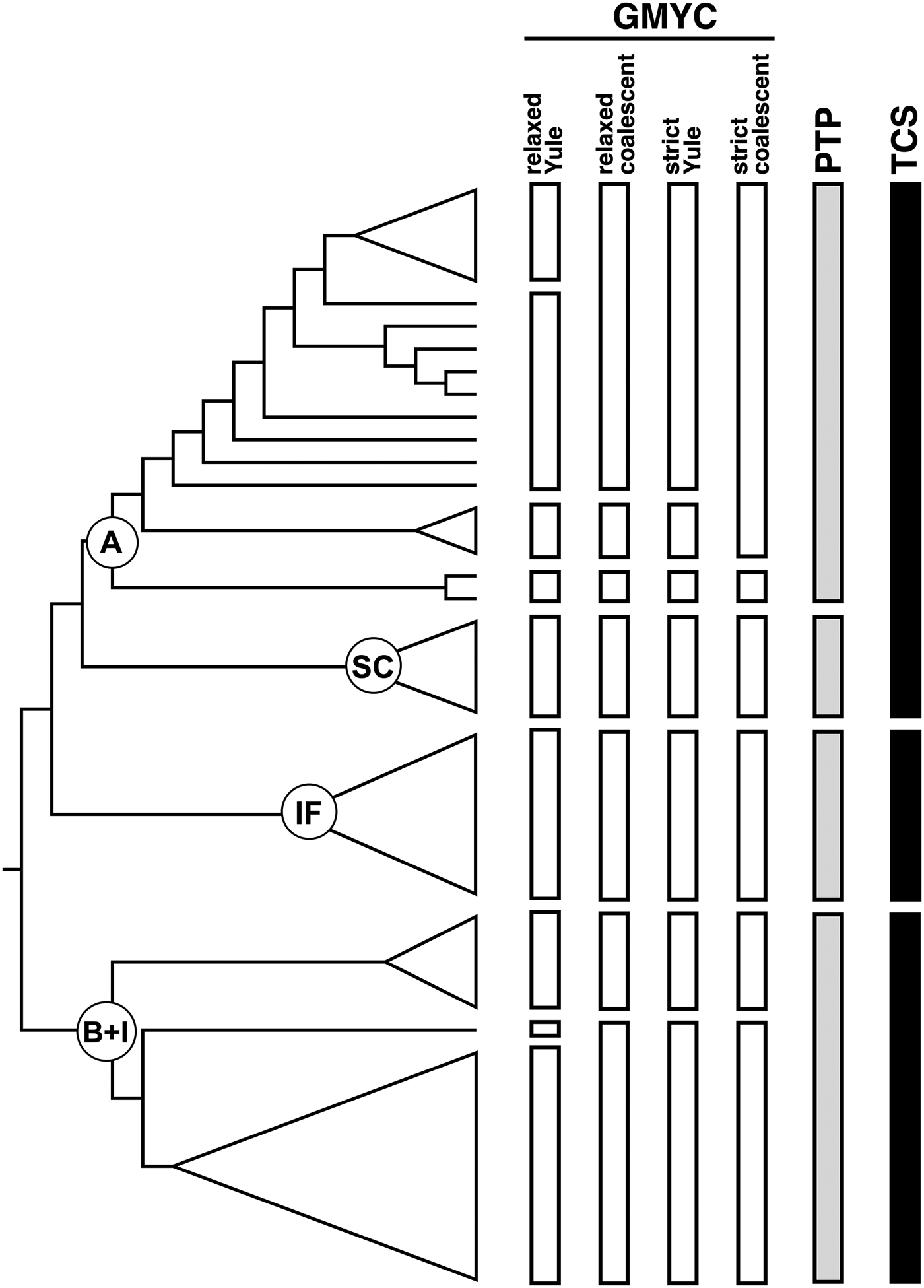
Overall, the three species delimitation methods are congruent in defining some entities and discordant in other instances (fig. 3). The GMYC method gave the higher number of putative entities, ranging from 6 to 9 depending on the ultrametric tree used. When using the relaxed clock with Yule prior, GMYC splits lineage A into four distinct taxa, while it indicated only two possible species when using the strict clock tree with coalescent prior. In comparison, PTP recognized a single entity. Although the parsimony network groups lineages A and SC in a single taxon, the latter lineage is always defined as a single, separate entity in the other analyses (fig. 3). Lineage IF is indicated as a distinct taxon by all methods, while variation in species delimitation can be observed across methods for the K. flavicollis lineage B + K. italicus clade (fig. 3). GMYC defined three or two taxa and, again, the use of relaxed clock with Yule prior gave more estimated species. On the other hand, PTP and parsimony analyses indicated this clade as a single entity.
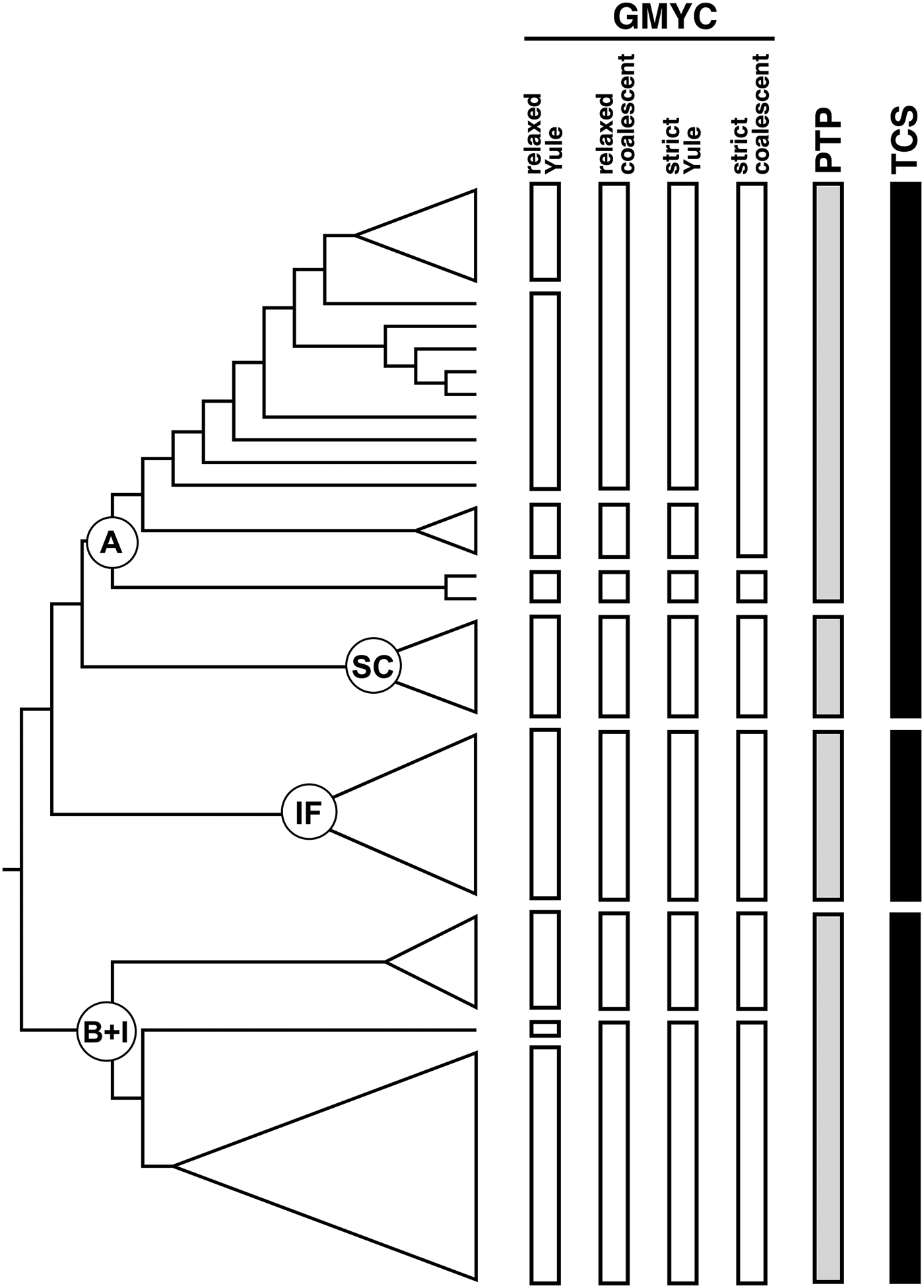
Fig. 3. Species delimitation analyses. Main clades are indicated by circles at their respective ancestral nodes. Outgroups have been omitted in the figure. Abbreviations: A, lineage A; SC, Sardo-Corsican lineage; IF, Ibero-French lineage; B + I: lineage B and Kalotermes italicus.
Of the 38 colonies for which two individuals were sequenced, different haplotypes were found in ten (26.3%; table 1). In six instances, the two distinct haplotypes even belong to different clusters (table 1; fig. 2; summarized in table 3). The Sicilian sample from Cinnisi (CNS) exhibited haplotypes from K. flavicollis lineages A and SC, while two San Rossore colonies (ROSb and ROSc) carried haplotypes from K. flavicollis lineage SC and K. flavicollis lineage B + K. italicus clade. Finally, colonies FENb and FENc, from the Feniglia Natural Reserve, and SIRa, from Sirolo, contained haplotypes of both K. flavicollis lineage A and K. flavicollis lineage B + K. italicus clade.
Table 3. Colonies with mixed haplotype composition.

Discussion
The evolutionary diversification pattern of the genus Kalotermes is poorly known in Europe, compared with the European Reticulitermes. In particular, the taxonomic level of divergence among lineages and the geographical range of taxa distribution still remain to be defined. The present survey provides additional knowledge on the systematics, evolutionary history, and biogeography of Kalotermes taxa across the Mediterranean area.
Phylogenetic relationships among Kalotermes lineages
The present analysis is based on a single mitochondrial fragment that proved to be informative, especially to identify new phylogenetic lineages (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011; Ghesini & Marini, Reference Ghesini and Marini2013, Reference Ghesini and Marini2015; Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a,Reference Luchetti, Scicchitano and Mantovanib). The nucleotide variability scored reveals that the Kalotermes genus in Western Europe is structured in two well-supported clusters. The first one, including lineages A, SC, and IF, clearly shows a monophyletic origin, with lineage IF branching first. The relationship between lineages A and SC appears less clear, mostly due to Lussino and Portoscuso haplotypes, which clearly diverged from lineage A. The colony of Portoscuso was already interpreted as a divergent haplotype within lineage A (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011). The second cluster includes sequences of K. flavicollis lineage B (Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a) and the recently described species K. italicus (Ghesini & Marini, Reference Ghesini and Marini2013). This cluster is partitioned in two sub-clusters, with a nucleotide divergence similar to the one scored between K. flavicollis lineages A and SC (1.2 vs. 1.5%; table S1). However, haplotype pairs belonging to lineage B and K. italicus samples cluster together, supporting the hypothesis that K. flavicollis lineage B and K. italicus are the same taxon. Therefore, all samples included into this cluster will be considered as K. italicus.
Species delimitation and taxonomic considerations
The three methods used to delimitate Kalotermes species gave slightly different results. The GMYC method, which is widely used for species delimitation on a single marker, is known to be strictly dependent on the algorithm used for ultrametric tree calculation (Monaghan et al., Reference Monaghan, Wild, Elliot, Fujisawa, Balke, Inward, Lees, Ranaivosolo, Eggleton, Barraclough and Vogler2009; Tang et al., Reference Tang, Humphreys, Fontaneto and Barraclough2014). The analysis conducted with Kalotermes sequences confirmed this observation, with different results depending on the clock model and/or the tree prior used. The use of a strict clock with a coalescent prior gave the most conservative result and it is more consistent with PTP and parsimony analyses. Irrespective of the clock model and prior used, GMYC analyses always indicated that Portoscuso and Lussino haplotypes constitute a taxonomic entity that is separated from all other haplotypes grouped within lineage A. This is consistent with previous results (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011). On the other hand, the PTP and parsimony analyses did not differentiate these two haplotypes from other clades within lineage A. It has been observed that the GMYC method may not perform well when dealing with poly- or paraphyletic lineages (Hendrich et al., Reference Hendrich, Pons, Ribera and Balke2010): this could be the case of K. flavicollis lineage A, as the divergence of Portoscuso and Lussino haplotypes place them in an unresolved position (fig. 2).
On the whole, lineages A and SC most likely represent two distinct taxonomic entities within K. flavicollis, even if the parsimony analysis group them together. These results are in line with Springhetti's preliminary studies, which found differences of morphometric parameters and reproductive traits between Sardinian and Italian peninsular colonies (Springhetti, Reference Springhetti1967). The K. flavicollis lineage IF is consistently recognized as a single, separate taxon; as previously found (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011), molecular data mirror the physiological divergence observed by Luscher (Reference Luscher1956) between Italian and French Kalotermes populations. This suggests that K. flavicollis lineage IF might represent a new Kalotermes species. Except for GMYC analysis, the two other analyses indicated that K. italicus most likely constitute a single taxon, even if it is structured into two sub-clusters which might reflect some degree of intraspecific differentiation.
Biogeography and evolution of European Kalotermes termites
Data presented in this study revealed a significant phylogeographic structure of western European Kalotermes termites. The phylogenetic relationships among K. flavicollis lineages are indeed consistent with their geographic location (summarized in fig. 1b). Present study supports a wider distribution of K. flavicollis sensu stricto (lineage A), its range spanning from the Aegean coasts to the whole Italian Peninsula and Sicily. Our results also revealed that K. flavicollis lineage SC is not restricted to Sardinia and Corsica islands, as previously thought (Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011), but it is also present on the mainland, along Ligurian and Tuscanian coasts.
The phylogeographic pattern found in Kalotermes lineage SC nicely mirrors that observed in Reticulitermes lucifugus subspecies, with the Sardo-Corsican R. lucifugus corsicus observed also on the mainland (Luchetti et al., Reference Luchetti, Scicchitano and Mantovani2013b). R. lucifugus diverged from the Iberian lineage and migrated to the Sardo-Corsican microplate after its detachment from the Iberian Peninsula (~10 million years ago; Dedeine et al., Reference Dedeine, Dupont, Guyot, Matsuura, Wang, Habibpour, Bagnères, Mantovani and Luchetti2016). Although our analyses do not provide time estimates, K. flavicollis sensu stricto and lineage SC could have followed a similar path. In fact, its close relationship with the lineage IF cluster is reminiscent of the relationship between Iberian Reticulitermes grassei -Reticulitermes banyulensis and the R. lucifugus corsicus subspecies (Luchetti et al., Reference Luchetti, Scicchitano and Mantovani2013b; Dedeine et al., Reference Dedeine, Dupont, Guyot, Matsuura, Wang, Habibpour, Bagnères, Mantovani and Luchetti2016). Although the dataset might be limited, it is interesting that K. flavicollis sensu stricto and lineage SC show signatures of a recent and rapid population growth (Tajima's Ds −2.227 and −2.413, P < 0.01), while lineage IF does not. This pattern possibly results from Pleistocenic glaciations, which could have imposed a southward contraction of the Italian population, followed by a recolonization after climate warming (Hewitt, Reference Hewitt1996). On the contrary, lineage IF appears to have remained in equilibrium, suggesting the possibility that it was not affected by Quaternary climatic oscillations. Still, the Tajima's D value obtained with Ibero-French lineage was negative, suggesting that this lineage may have experienced a more limited population expansion.
The distribution of K. italicus is limited to certain areas along the northern Tyrrhenian coast, overlapping the northern edge of K. flavicollis sensu stricto distribution, and in two areas on the Adriatic side. This can be explained either by a naturally limited distribution or by a more recent colonization from an unknown area. Our analyses showed that K. italicus is genetically structured and does not exhibit any signature of population size changes. In fact, the Tajima's D value is not significantly different from 0, suggesting that K. italicus is at mutation-drift equilibrium. The high genetic diversity of this species might suggest that K. italicus geographical range is rather stable, although such an hypothesis remains to be tested. However, recent colonizations by this species seem rather unlikely since such events usually result in population bottlenecks. An alternative explanation is that K. italicus was introduced several times in the same places. Termites are indeed easily transported by means of human activities, for instance through lumber industry and/or wooden artifacts trade (Evans et al., Reference Evans, Forschler and Grace2013; Scicchitano et al., Reference Scicchitano, Dedeine, Bagnères, Luchetti and Mantovani2017), sometimes confounding the study of natural distributions. In order to precisely determine the natural distribution of these organisms, a large and detailed sampling is often required (Luchetti et al., Reference Luchetti, Scicchitano and Mantovani2013b).
Interspecific colony fusion and implications for hybridization
Three types of colony breeding structure are known in termites: (i) simple families are composed of offspring from a primary couple; (ii) extended families possess offspring of primary and/or secondary reproductives; (iii) mixed families include offspring of more than two unrelated reproductives (Vargo & Husseneder, Reference Vargo, Husseneder, Bignell, Roisin and Lo2011). Nearly one-third of the presently analyzed colonies are mixed families exhibiting two distinct haplotypes, indicating that at least two females are involved in the reproduction (table 3). Mixed-family colonies are not rare in termites, especially in termopsid and kalotermitid species: in these taxa, several studies showed that independent colonies of the same taxon can fuse into a single social entity (Thorne et al., Reference Thorne, Breisch and Muscedere2003; Johns et al., Reference Johns, Howard, Breisch, Rivera and Thorne2009; Velonà et al., Reference Velonà, Luchetti, Ghesini, Marini and Mantovani2011; Korb & Roux, Reference Korb and Roux2012; Howard et al., Reference Howard, Johns, Breisch and Thorne2013; Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a). We recently reported an extreme case of colony fusion in an Italian population of K. flavicollis (Feniglia Natural Reserve; Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a) with an exceptionally high frequency of mixed-family colonies, containing up to nine mitochondrial haplotypes. That study found also that some mixed-family colonies contained haplotypes belonging to the two divergent lineages A and B, which are here assigned to K. flavicollis sensu stricto (lineage A) and K. italicus (lineage B), respectively. In the present analysis, we found three further mixed-family colonies showing K. flavicollis sensu stricto and K. italicus haplotypes. For the first time, we also found two mixed-family colonies with K. flavicollis lineage SC and K. italicus haploypes and another one with K. flavicollis sensu stricto and K. flavicollis lineage SC haplotypes. These new results suggest that also interspecific colony fusion could be a widespread phenomenon in Kalotermes taxa.
It is interesting to consider possible outcomes of interspecific colony fusion. In the previous study, mixed-family colonies of K. flavicollis sensu stricto/K. italicus (at that time only indicated as lineages A and B, respectively; Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a), the analysis of nuclear microsatellite markers indicated that individuals with K. flavicollis sensu stricto mitochondrial haplotype showed nuclear genetic membership to K. italicus and vice-versa. This indicated that the two taxa are able to interbreed (Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a), thus suggesting that K. flavicollis sensu stricto and K. italicus may naturally hybridize. When Ghesini & Marini (Reference Ghesini and Marini2013) described K. italicus species they proposed that, based on morphological evaluations, the taxon K. flavicollis var. fuscicollis observed by Becker (Reference Becker1955) might be the result of K. flavicollis sensu stricto and K. italicus hybridization. Interestingly, Becker (Reference Becker1955) himself showed that Kalotermes individuals with black pronotum and K. flavicollis sensu stricto may interbreed, also giving viable offspring.
Species hybridization in social insects is not expected to occur at a high rate, but it was, nevertheless, evidenced in ants and termites (Feldhaar et al., Reference Feldhaar, Foitzik and Heinze2008). In termites, instances of natural hybridization and/or introgression were observed in lower termites, such as Zootermopsis and Kalotermes (Aldrich & Kambhampati, Reference Aldrich and Kambhampati2007; Luchetti et al., Reference Luchetti, Dedeine, Velonà and Mantovani2013a), and Rhinotermitidae (Coptotermes spp. and Reticulitermes spp.; Lefebvre et al., Reference Lefebvre, Châline, Limousin, Dupont and Bagneres2008; Chouvenc et al., Reference Chouvenc, Helmick and Su2015; Lefebvre et al., Reference Lefebvre, Vargo, Zimmermann, Dupont, Kutnik and Bagnéres2016). Moreover, laboratory colonies established by heterospecific mates in Nasutitermes corniger × Nasutitermes ephratae and Coptotermes formosanus × Coptotermes gestroi pairs were found to be more productive in term of offspring output (Hartke & Rosengaus, Reference Hartke and Rosengaus2011; Chouvenc et al., Reference Chouvenc, Helmick and Su2015). It is still not clear if the high frequency of colony fusion observed in Kalotermes might have facilitated the hybridization or if it is the reverse situation. Further studies along the sympatry area between K. flavicollis and K. italicus would likely provide interesting insight into reproductive boundaries and colony mate recognition in these social insects.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0007485317001080
Acknowledgements
This work was supported by RFO-UNIBO and Canziani funding to AL and BM. The authors would like to thank Claudia Scavariello, Davide di Domenico, Lara Maistrello, and Simone Marchetti for providing samples. AL is particularly grateful to Nicola and Nicoletta Ciarelli for they let him sample termites during their wedding party. Finally, the authors wish to thank three anonymous Reviewers and the Editor for their thorough revision and helpful comments.