Introduction
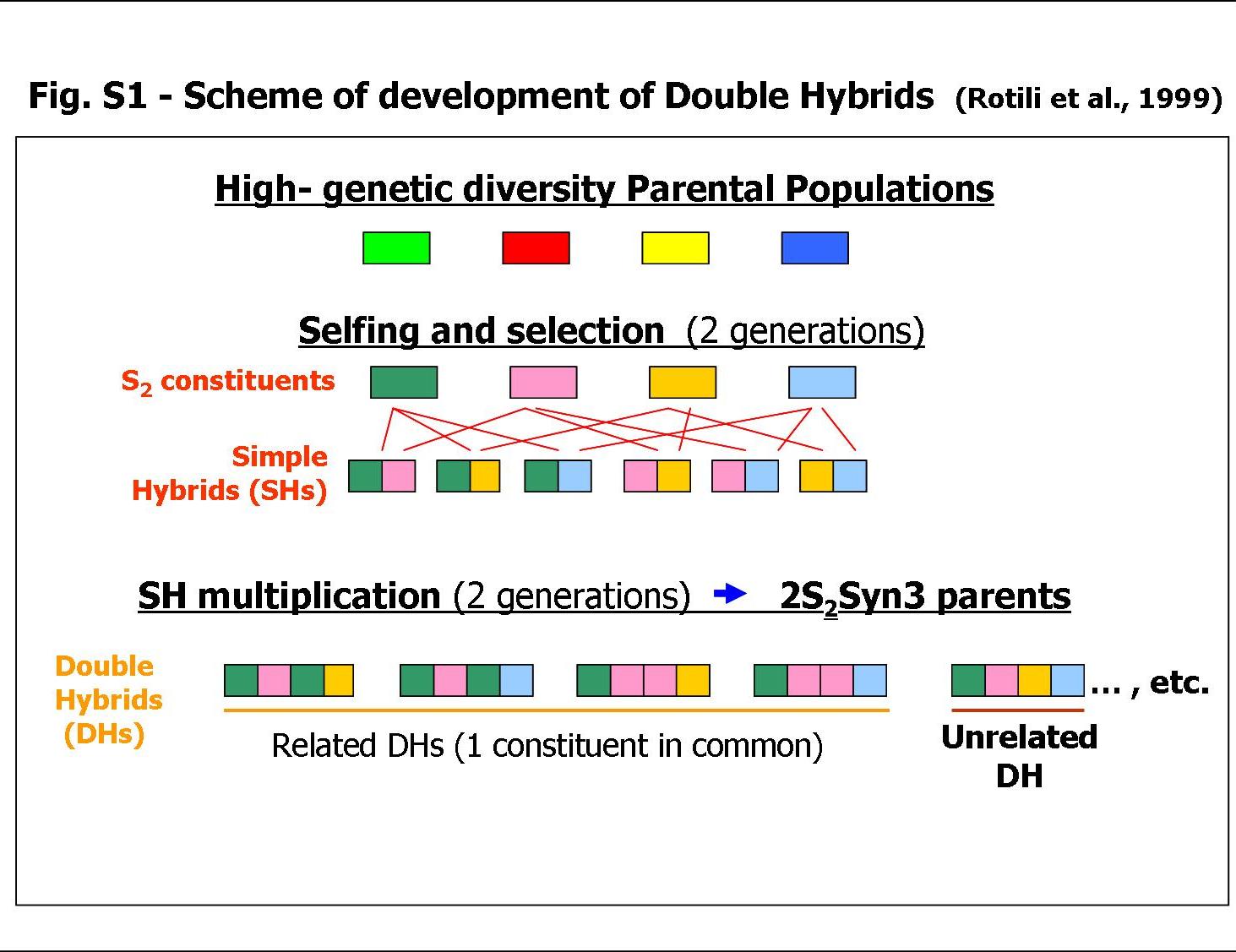
Double ‘free-hybrids’ (DH, obtained in the absence of male sterility mechanisms and consequently with a same theoretical probability of within- and among-population crossings) in alfalfa, an autotetraploid, allogamous and perennial forage crop, were obtained using a variety construction process developed at the Lodi Institute (Rotili et al., Reference Rotili, Gnocchi, Scotti and Zannone1999; see Supplementary Fig. S1, available online only at http://journals.cambridge.org).
The aim of this study was to analyze the heterosis effect in alfalfa by comparing the DH and their respective parents (multiplied simple hybrids (SHs) or 2S2Syn3) using two molecular approaches: first, by estimating the genetic diversity and the heterozygosity levels using simple sequence repeat (SSR) molecular markers and, second, by analyzing the variation in gene expression using microarrays.
Materials and methods
Plant material
S2 families originating from eight unrelated and geographically distant populations, selected for dry matter production (DMY) during selfing, were crossed manually without emasculation to produce SH in a diallelic scheme. One single plant/family was used for each cross (seven plants/family in total). The six SHs from the four parents with the highest general combining ability (GCA) effects were multiplied upto the Syn3 generation and finally crossed in a diallelic scheme to produce DH. DHs were produced in different years, by two procedures: using a single multiplied SH plant selected for vigour, for each parent (five plants/family in total) or crossing four plants/parental SH (20 plants/family in total). The DHs, and their respective parents, obtained with the second procedure were used for SSR analysis.
Two diallelic crosses (diallel A and B), 6 × 6, were obtained using the above process and the resulting DHs were analyzed for DMY in greenhouse conditions for 2 years (ten harvests) together with their parents (150 plants/family at the density of 250 plants/m− 2). All the 15 DHs of diallel A were analyzed using the SSR approach (ten plants/DH family, five vigorous and five weak; 150 plants in total).
Molecular analysis
Sixty-three microsatellite (SSR) markers derived from both M. truncatula and M. sativa were analyzed by multiplex PCR reactions and capillary electrophoresis in an automated detection system (ABI 310; Applied Biosystems, USA). Heterozygosity was estimated by means of the average number of alleles (peaks)/locus.
Microarray analysis
The heterotic DH B2 × B5 line was chosen for transcriptome analysis. Both the DH forms, obtained using either one or four plants/parent, were grown and displayed a similar heterotic behaviour. The microarray analysis was performed on the first form. The transcriptome of the DH B2 × B5 was compared with the two parents (2S2Syn3) using the Affymetrix Medicago Genome Array (Affymetrix, USA). RNA was extracted from leaves of plants at early flowering stage, each parent of the DH B2 × B5 being a single plant; the RNA of ten heterotic progenies of the DH was bulked in one sample. Hybridization signals were analysed using the Genespring GX11 software (Agilent technologies, USA) using robust multichip average (RMA) prenormalization algorithm. Subsequently per-gene normalization was performed by standardizing the probe set signals to the median value for all arrays. Gene expression was classified as additive/non-additive according to Hochholdinger and Hoecker (Reference Hochholdinger and Hoecker2007). Genes having a hybrid expression value similar to the parental average (fold change < 1.5, t-test P ≤ 0.05) were considered as additive. Non-additive genes were classified in functional categories using the GeneBins software (Goffard and Weiller, Reference Goffard and Weiller2007). All the data have been donated to the GEO database (http://www.ncbi.nih.gov/geo/, with accession ID GSE25034).
Results and discussion
Diallelic analysis indicated specific combining ability (SCA) as the main source of variation for DMY in the DHs (Fig. 1). This important deviation from additivity expressed by SCA supported heterosis values versus the best parent of +50% in the DH B2 × B5 and an average of +45% (ranging from +5 to +76%) in the 15 DHs of diallel A. The diallel hybridization of six SHs, from the diallel crossing of four S2 lines, produced unrelated (3 out of 15) and related (12 out of 15) DHs, based on the presence or absence of a constituent in common between the two parental SHs. The genetic similarity between parents, estimated by Dice coefficient based on 63 SSR markers, was consistent with the kinship degree of parents. In diallel A, the related parents showed an average similarity of 0.61, significantly higher than the value of 0.56 found for the three unrelated DHs. In diallel B, the genetic similarity between the related parental SHs B2 and B5 was 0.64. Parental genetic diversity showed a significant relationship with DMY (r = 0.59; P < 0.05), heterosis versus the best parent (r = 0.70; P < 0.01) and SCA effects (r = 0.76; P < 0.005) of DH progenies in diallel A.

Fig. 1 Diallel analysis: variance partitioning for DMY in diallel crosses.
Parental diversity was the basis of the significant heterozygosity recovery in DHs with respect to the parents, 2.16 alleles/locus versus 2.11 in parents in diallel A (average of 15 DHs and their respective parents) and 2.14 versus 2.04 in B2 × B5. However, the variation of heterozygosity estimates explained only a small part (~20%) of the variation in DMY of the DHs (r = 0.45; P>0.05, in diallel A), while the number of alleles based on all the 63 SSR loci (allelic richness) was significantly related to DM performance (r = 0.61; P < 0.05).
It should be feasible to relate SCA values, found in the diallelic analysis of DHs to different genetic parameters including allelic interactions, estimated by heterozygosity level/locus; non-allelic interactions, estimated by allelic richness on the whole set of SSR loci studied; positive complementation of specific interactions, estimated by the study of differential gene expression of a DH and the respective parents.
Expression analysis of the heterotic hybrid B2 × B5 revealed that 87% of the probe sets were expressed in an additive manner (Fig. 2(a)). Of the genes expressed non-additively, the majority (86%) were outside the parental range, i.e. either above the high parent or below the low parent expression levels (Fig. 2(b)). To confirm these results, gene expression values were validated for 14 out of 17 selected genes using quantitative-PCR (data not shown).

Fig. 2 Transcriptome analysis. (a) Proportions of genes falling in the additive/non-additive category. (b) Classification of genes assigned to the non-additive category.
To further investigate the function of these gene sets, gene ontology analysis was performed. Analysis of the genes expressed outside the parental range showed that the highest represented categories (excluding unclassified and without homologue, 44.1 and 13.9%, respectively) were metabolism (carbohydrate 10.5%; amino acid 7.3%; cofactors and vitamins 6.3%; lipid 5.6%; nucleotide 4.9%; secondary metabolites 4.8%) and genetic information processing (signal transduction 7.2%; translation 6.0%; folding, sorting and degradation 6.1%). Equivalent additive gene expression has been observed in other studies of heterotic hybrids in different species (maize, Arabidopsis; reviewed by Hochholdinger and Hoecker, Reference Hochholdinger and Hoecker2007). Interestingly, Li et al. (Reference Li, Wei, Nettleton and Brummer2009), studying free-hybrids between a Medicago falcata and two M. sativa populations, reported that probe sets exhibiting expression levels different from midparent value (non-additive expression) had a higher proportion displaying over/under-dominance in heterotic (interspecific hybrids, i.e. falcata × sativa) than in non-heterotic (intraspecific, i.e. sativa × sativa) hybrids. Both experiments indicate that genes with non-additive expression, and in particular those exhibiting over/under dominance, play a role in yield heterosis in alfalfa. It is worth underlining that, in an autotetraploid allogamous species such as alfalfa and in the free-hybrid construction process used, the parental multiplied SHs are not homozygous as the maize inbred lines, but have, on average, 2.11 (diallel A) alleles/locus. As a consequence, parental performance, either agronomic (DMY) or as gene expression, is in part based on interactions between the two different alleles and deviations from additivity (SCA in the case of DMY and over/under dominance in the case of gene expression) reflect mainly higher order interactions at single loci and/or epistatic phenomena.
A combination of agronomic and advanced molecular approaches is likely to have great potential in highlighting the genetic basis of heterosis in autotetraploids species.