


Introduction
Efforts to protect rare species, manage pests or invasive species, and control diseases require the accurate identification of the target species. Similarly, biodiversity assessments also necessitate reliable discrimination among related species. In the past two decades, it has become very clear that cryptic species, i.e. genetically distinct but morphologically similar species, are extremely common among all higher taxa (Bickford et al., Reference Bickford, Lohman, Sodhi, Ng, Meier, Winker, Ingram and Das2007). When correcting for study effort and accounting for other confounding variables, cryptic species do not appear to be more frequent among parasitic than free-living taxa (Poulin & Pérez-Ponce de León, Reference Poulin and Pérez-Ponce de León2017). Yet cryptic parasite species are reported with increasing frequency; this is a consequence of genetic methods having become standard practice for parasite identification, and often being used deliberately to prospect for cryptic diversity (Blouin, Reference Blouin2002; Locke et al., Reference Locke, McLaughlin and Marcogliese2010; Rosas-Valdez et al., Reference Rosas-Valdez, Choudhury and Pérez-Ponce de León2011; Razo-Mendivil et al., Reference Razo-Mendivil, Rosas-Valdez, Rubio-Godoy and Pérez-Ponce de León2015). For parasitologists, there are serious implications: the occurrence of cryptic parasite species can greatly complicate parasite taxonomy, bias estimates of host specificity and undermine our efforts to control parasitic diseases (Poulin & Keeney, Reference Poulin and Keeney2008; Pérez-Ponce de León & Nadler, Reference Pérez-Ponce de León and Nadler2010; Nadler & Pérez-Ponce de León, Reference Nadler and Pérez-Ponce de León2011).
We still have a limited understanding of the distribution of cryptic parasite diversity, and whether it is more widespread in certain taxa than others. If we overlook whether they are free-living or parasitic, reports of cryptic species are more common in certain higher taxa (phyla or classes) of animals, and less common in others, than expected based on their known species richness and on research effort (Pfenninger & Schwenk, Reference Pfenninger and Schwenk2007; Pérez-Ponce de León & Poulin, Reference Pérez-Ponce de León and Poulin2016). This suggests an uneven distribution of cryptic diversity among higher taxa. A few years ago, Poulin (Reference Poulin2011) reported a similar uneven distribution among higher groups of parasitic helminths. Specifically, after correcting for sampling effort (i.e. number of parasite individuals from which sequence data are obtained), more cryptic species of trematodes are found per published study than among other helminth taxa, namely cestodes, monogeneans, nematodes and acanthocephalans (Poulin, Reference Poulin2011). However, many studies reporting cryptic parasite species have been published in the past few years, providing a much larger dataset to re-examine this finding in the light of new evidence.
Here, we re-visit Poulin's (Reference Poulin2011) observation that cryptic species are more common among trematodes than other helminths, using an expanded dataset including almost three times more studies than the 33 that were available for Poulin's (Reference Poulin2011) analysis. Our results confirm that, all else being equal, more cryptic species tend to be found per study effort among trematodes than other groups of parasitic helminths, an intriguing pattern for which we propose some explanations.
Materials and methods
We used the dataset of Poulin & Pérez-Ponce de León (Reference Poulin and Pérez-Ponce de León2017), based on an exhaustive search of the literature in the main collection of the ISI Web of Science™ for the period 1978–2015, and extended it up to June 2016. The updated dataset compiles data from published studies on cryptic species, resulting from a search using the terms: ‘cryptic speci*’ OR ‘cryptic linea*’ OR ‘cryptic tax*’ OR ‘sibling species’ in either the title, abstract or keywords of papers. The entries recovered were then reduced to only those pertaining to Trematoda (= Digenea), Cestoda, Monogenea, Acanthocephala and Nematoda (parasitic taxa only, with those parasitic on animals and plants treated separately). Each record was checked individually to eliminate non-relevant articles, and we retained only true reports of cryptic species using DNA sequence data to discover or recognize cryptic species. We included both explicit prospecting studies deliberately searching for cryptic species, and other studies that discovered cryptic species as a by-product of genetic analyses with other purposes. We also retained studies where an attempt was made to identify cryptic species using sequence data, but in which no cryptic species were actually found.
We use the following operational definition of ‘cryptic species’: genetically distinct taxa previously not recognized and first uncovered through genetic analysis. Different authors use different levels of genetic difference (i.e. % base pair differences) and different approaches to identify a taxon as a ‘cryptic species’ distinct from already known species. Here, we accepted the expertise of the authors of the original studies and used their estimate of the number of cryptic taxa in their samples. The final dataset includes 110 studies, published between 1995 and 2016 (supplementary table S1).
For each study, we recorded: (1) the species, genus or family targeted by the study; (2) the taxon to which it belonged, i.e. Trematoda, Cestoda, Monogenea, Acanthocephala, animal-parasitic Nematoda and plant-parasitic Nematoda; (3) the habitat in which it is found, either predominantly terrestrial, freshwater or marine; (4) the biogeographical region where the study was conducted, i.e. Afrotropical, Australasian, Boreal, Indo-Malayan, Nearctic, Neotropical, Palaearctic or more than one of them; (5) the numbers of mitochondrial markers, sequences and base pairs used; (6) the numbers of nuclear markers, sequences and base pairs used; and finally (7) the number of cryptic species detected beyond the originally known species (see supplementary table S1).
The number of sequences represents the sum of sequences obtained across different markers, including multiple copies of the same haplotype or genotype, calculated separately for mitochondrial and nuclear genes; it captures both the number of markers and the number of individuals from which DNA was obtained, and is used here as a measure of sequencing effort instead of the number of individuals, since the latter varies across markers. Also, the number of markers, the number of sequences and the number of base pairs generally correlate positively with each other across studies, for both mitochondrial and nuclear genes (see Poulin & Pérez-Ponce de León, Reference Poulin and Pérez-Ponce de León2017). Therefore, we use only the number of sequences to control for study effort in subsequent analyses, since the number of cryptic species found tends to increase with the level of effort put into their search (Poulin, Reference Poulin2011).
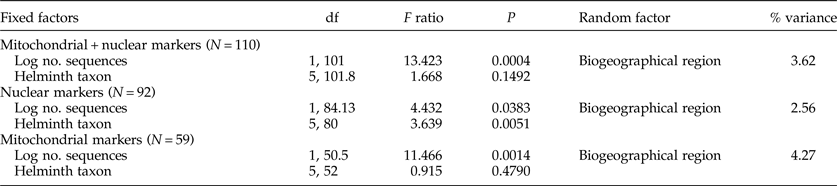
We tested the determinants of the number of cryptic species found per study (response variable) using generalized linear mixed models with Poisson error structure. The fixed factors were helminth taxon (Trematoda, Cestoda, Monogenea, Acanthocephala, animal-parasitic Nematoda and plant-parasitic Nematoda) and the log-transformed number of sequences obtained as a measure of study effort. The habitat in which the parasites occur (terrestrial, freshwater or marine) was also included as a fixed factor in initial models, but removed in the final models as its effect was totally negligible. To account for possible spatial variation in the formation of cryptic species (Pfenninger & Schwenk, Reference Pfenninger and Schwenk2007), the biogeographical region where each study was conducted was included as a random factor. All analyses were conducted in JMP v. 11.0 (SAS Institute Inc., Cary, North Carolina, USA). We ran the above model on three subsets of the data: only studies using mitochondrial markers, only studies using nuclear markers and all studies. In the latter case, the number of sequences obtained was calculated as the sum of mitochondrial and nuclear sequences.
Results
Our dataset comprised 110 studies using sequence data to uncover cryptic species of helminth parasites (see supplementary table S1). Of these, 59 used mitochondrial markers, 92 used nuclear markers and 41 used both types of markers. Among studies using both types of markers, the number of mitochondrial sequences and the number of nuclear sequences were positively correlated (r = 0.465, N = 41, P = 0.0022). The majority of studies were on either trematodes or animal-parasitic nematodes (table 1). Overall, the studies considered here uncovered 246 cryptic species (range 0–18 per study).
Table 1. Summary of the number of studies in the dataset on each of the parasitic helminth taxon, also showing the range in the number of cryptic species found per study. Full dataset available in supplementary table S1.
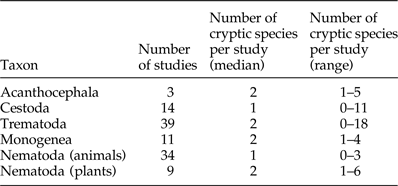
Whether we performed the analyses with only studies using mitochondrial markers, only studies using nuclear markers or all studies (summing up nuclear and mitochondrial sequences), the number of sequences obtained always emerged as a significant predictor of the number of cryptic species uncovered (table 2). The more sequences are obtained for a particular helminth taxon, the more cryptic species are found (fig. 1). The biogeographical region in which a study was conducted accounted for very little of the remaining variance in the number of cryptic species uncovered (table 2), whether we considered mitochondrial data only, nuclear data only or both.

Fig. 1. Number of cryptic helminth species found as a function of (a) the number of nuclear sequences obtained (N = 92) and (b) the total number of DNA sequences obtained (N = 110 studies), with different symbols for different helminth taxa. Nematodes parasitic in animals (A) and plants (P) are shown separately. The line represents the line of best fit from a linear regression.
Table 2. Results of the mixed-effects models with the number of cryptic helminth species found per study as response variable, showing the effects of the main predictors and the proportion of the remaining variance accounted for by the random factor. Models were run across all studies by computing the total number of sequences obtained (mitochondrial plus nuclear, including cases with zero sequences for either type of marker), and separately using only studies using mitochondrial sequences or only studies using nuclear sequences.
In contrast, the influence of the higher helminth taxonomic group to which the studied taxa belonged depended on whether we considered mitochondrial data only, nuclear data only or sequences of both types of marker combined (table 2). When we included only studies using nuclear markers, we found that more cryptic species tend to be uncovered among trematodes, and fewer among cestodes and animal-parasitic nematodes, than in other helminth groups when correcting for sequencing effort (fig. 2). The actual effect was small, i.e. approximately an extra two cryptic species found for an effort of 100 sequences in trematodes than for the same sequencing effort in cestodes, but significant. Roughly the same trend, though not significant, was also observed when the total number of sequences (nuclear plus mitochondrial) was used as a predictor in the analysis (fig. 2). Thus, for a given sequencing effort, more cryptic trematode species are likely to be revealed than for other helminth groups.
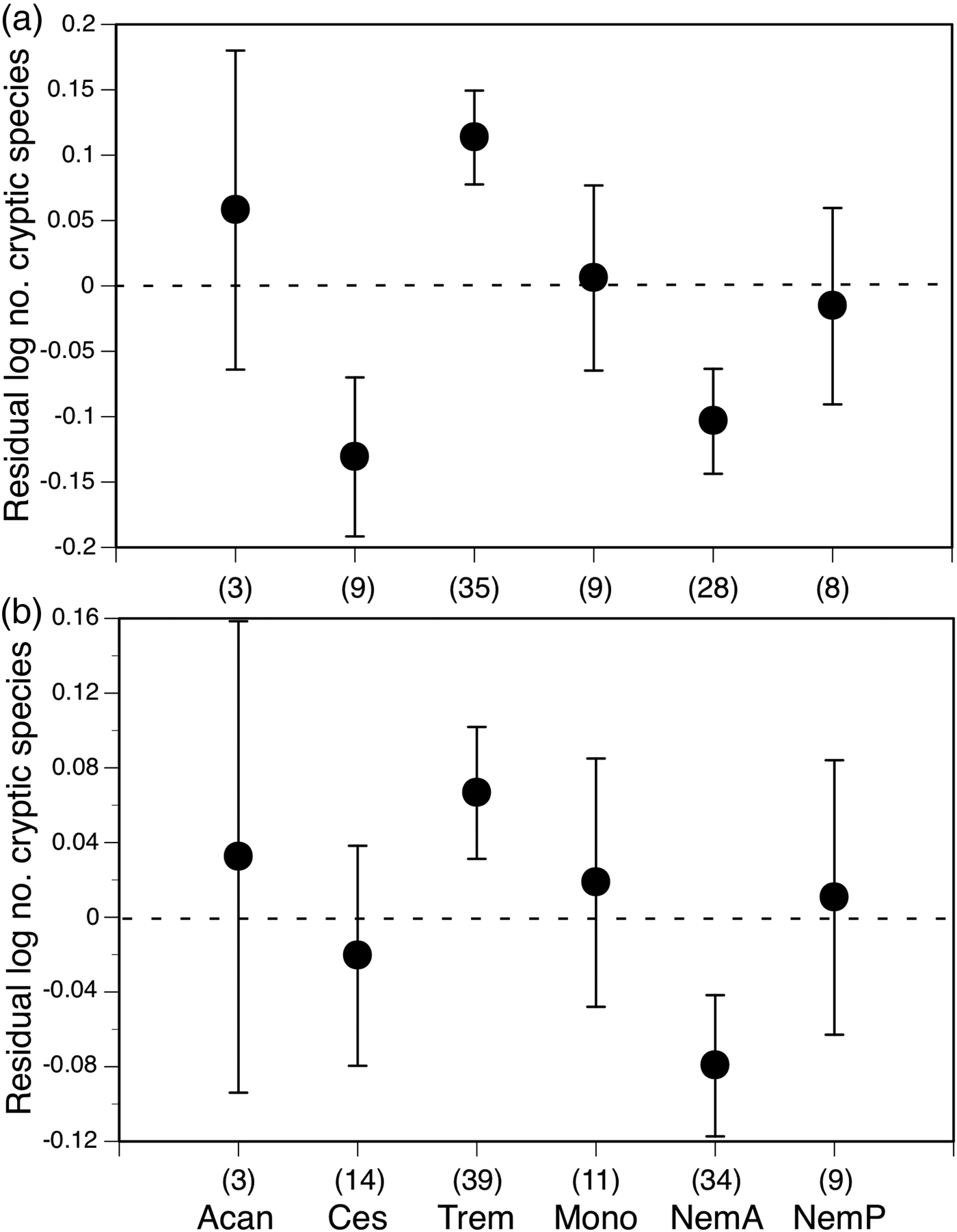
Fig. 2. Mean (± SE) residual number of cryptic helminth species found, from the regressions (in fig. 1) against (a) the number of nuclear sequences obtained and (b) the total number of DNA sequences obtained, shown separately for acanthocephalans (Acan), cestodes (Ces), trematodes (Trem), monogeneans (Mono), animal-parasitic nematodes (NemA) and plant-parasitic nematodes (NemP). Numbers of studies in each group are shown in parentheses.
Discussion
A few years ago, Poulin (Reference Poulin2011) reported that more cryptic species were uncovered among trematodes than other helminth groups when controlling for the number of DNA sequences obtained per study. Here, using a much larger dataset, we generally confirmed this earlier finding. There are several possible explanations for this pattern; we propose three distinct ones below.
First, there are intrinsic biological differences between trematodes and other parasitic helminths, and perhaps certain properties of trematodes promote the emergence of morphologically indistinguishable but genetically distinct lineages. In particular, unlike other helminths, trematodes undergo rounds of asexual multiplication within their molluscan first intermediate host. The possibility of somatic mutations occurring during this phase of asexual reproduction has been raised before as an explanation for slight genetic differences among cercariae issued from snails infected with a single miracidium (Yin et al., Reference Yin, Hu, Mo, Wang, Brindley, McManus, Davis, Feng and Blair2008). If this phenomenon is frequent, it could facilitate the rapid and common appearance of related species with very limited morphological differences.
Second, visual distinction between closely related trematode species may be more difficult because many trematode taxa lack fast-evolving hard structures, such as hooks, whose number, size and distribution serve as reliable and taxonomically informative morphological traits in other helminth groups (Vignon & Sasal, Reference Vignon and Sasal2010; Wayland, Reference Wayland2010). However, morphological differences among genetically distinct trematodes can be subtle, and mostly based on the size and distribution of some internal organs, usually associated with the reproductive system. In trematode families possessing hard structures, such as tentacle spines in the family Rhopaliasidae or oral collar spines in the family Echinostomatidae, morphological delimitation among species may be possible. In the Rhopaliasidae, this is sometimes accomplished by counting the number of spines in the retractile tentacles, or by measuring their size and distribution (Haverkost & Gardner, Reference Haverkost and Gardner2008). These morphological differences can be associated with genetically distinct lineages which seem to represent cryptic species in Rhopalias coronatus (López-Caballero, pers. com.). Hard, sclerotized structures are less likely to become distorted during preservation, and their general absence in most trematode families could make fine discrimination between similar species a little more problematic than for other groups of helminths. Still, in members of the Echinostomatidae, the number of spines in the oral collar is constant and when genetic differences are established among their members (e.g. Detwiler et al., Reference Detwiler, Zajac, Minchella and Belden2012; Georgieva et al., Reference Georgieva, Selbach, Faltýnková, Soldánová, Sures, Skírnisson and Kostadinova2013) trematode taxonomists have to look for other characters, either the size and distribution of these spines, or internal organs.
Third, it is possible that, for traditional reasons, the degree of morphological characterization of new species described by trematode taxonomists has not been quite as extensive as that seen in species descriptions of cestodes and nematodes. Indeed, based on a quantitative analysis of taxonomic quality among a large dataset on helminth species descriptions, Poulin & Presswell (Reference Poulin and Presswell2016) have found that descriptions of trematodes over the past few decades have consistently included fewer scanning electron micrographs than those of cestodes and nematodes. Coincidentally, cestodes and animal-parasitic nematodes are the two groups in which the fewest cryptic species are detected for a given sequencing effort, based on the present study. Nadler & Pérez-Ponce de León (Reference Nadler and Pérez-Ponce de León2011) actually consider cryptic species to be species that are difficult to recognize using traditional systematic methods. Therefore, perhaps slight morphological differences among related and sympatric trematode species are more likely to be missed upon initial description than for other helminth groups.
The other main finding of our study was that the number of DNA sequences obtained influences how many cryptic species are likely to be found, whatever their taxonomic group. Just as host sampling effort correlates strongly with how many species are detected in wildlife surveys (Walther et al., Reference Walther, Cotgreave, Price, Gregory and Clayton1995), we found that sequencing effort had a strong effect on how many cryptic helminth species were found per study. This supports earlier findings on different datasets (Poulin, Reference Poulin2011; Poulin & Pérez-Ponce de León, Reference Poulin and Pérez-Ponce de León2017). In the period covered by our study (1995–2016), the number of parasite sequences obtained per study has increased modestly but significantly over time (log-transformed sequence data, total: r = 0.354, N = 110, P = 0.0002; mitochondrial only: r = 0.251, N = 59, P = 0.0552; nuclear only: r = 0.340, N = 92, P = 0.0009). Therefore, there are encouraging signs that the decreasing cost of gene sequencing is no longer constraining sequencing effort. This fact, combined with the increasing sophistication of methods of species discrimination using DNA sequences (e.g. Puillandre et al., Reference Puillandre, Lambert, Brouillet and Achaz2012; Carstens et al., Reference Carstens, Pelletier, Reid and Satler2013; Flot, Reference Flot2015), means that near-future studies will be unlikely to miss cryptic species due to inadequate search effort.
Interestingly, the main result here, i.e. that more cryptic species tend to be found among trematodes than other helminth groups for a given sequencing effort, was only significant when we restricted the analysis to nuclear sequences. The trend was apparent but non-significant when all sequences were used, and not seen at all when only mitochondrial sequences were used. This is a striking result and at the moment we cannot explain it because it has been shown that mitochondrial DNA is the marker of choice to search for potential cryptic species in molecular prospecting studies of parasites (Blouin, Reference Blouin2002; but see Galtier et al., Reference Galtier, Nabholz, Glemin and Hurst2009). Blouin (Reference Blouin2002) and Vilas et al. (Reference Vilas, Criscione and Blouin2005) examined the relative merits of mitochondrial and nuclear genes for their use in prospecting for cryptic species in parasitic nematodes and platyhelminths, and demonstrated that mitochondrial DNA increases the probability of detecting diagnostic characters between cryptic species, because these genes possess a higher rate of evolution and smaller effective population size. However, mitochondrial DNA has not been used in all parasitic helminths on a regular basis to detect cryptic species; for instance, in parasitic nematodes, of the 43 cryptic species reports (see supplementary table S1), 28 used nuclear markers exclusively to detect cryptic species, and seven used only a mitochondrial gene. In contrast, in parasitic platyhelminths (all combined), only 10 used nuclear genes exclusively, and 21 used only mitochondrial genes. In this group, the trend is to use a combination of molecular markers since half of the 64 cryptic species reports used both nuclear and mitochondrial markers to detect cryptic species. Just for trematodes, Blasco-Costa et al. (Reference Blasco-Costa, Cutmore, Miller and Nolan2016) recently found that in the past 5 years most of the studies focusing on taxonomy, diversity, phylogeny and life cycles that generated mitochondrial sequences also reported nuclear DNA sequences. Actually, Blasco-Costa et al. (Reference Blasco-Costa, Cutmore, Miller and Nolan2016) argued that the best practice for the discipline would be to obtain sequence data from at least two independently evolving or unlinked loci, i.e. one mitochondrial and one nuclear. In any event, one possible reason for the pattern observed in the present study is that the analysis restricted to mitochondrial sequences was based on fewer studies than analyses of either nuclear markers or both markers combined (59 versus 92 or 110, respectively). Another possible reason might be differences in sequencing effort between studies using nuclear markers (N = 92; mean ± SE number of sequences obtained: 54.1 ± 7.7) and those using mitochondrial markers (N = 59; 82.8 ± 9.6). However, although the difference was significant, sequencing effort was generally greater for mitochondrial markers than for nuclear ones (t 149 = 2.34, P = 0.0204), and therefore discrepancies in sequencing effort cannot account for the lack of effect in our analysis when only mitochondrial sequences were used.
In conclusion, we confirm Poulin's (Reference Poulin2011) earlier finding that, for a given search effort, more cryptic species are uncovered among trematode taxa than for other groups of parasitic helminths, at least when using nuclear markers. Although the reasons remain unclear, the implications are important. Although these numbers are likely inaccurate, estimates of global trematode diversity are slightly higher than those for cestodes and animal-parasitic nematodes, much higher than those for acanthocephalans, and lower than those for monogeneans (Poulin, Reference Poulin2007). Based on our findings, it is likely that current estimates of regional or global trematode diversity are relatively further from the true diversity than corresponding estimates for other helminth groups, where cryptic diversity is less widespread. Our results also suggest that the task ahead for trematode taxonomists may be huge, because many species requiring proper characterization probably remain hidden within previously described taxa.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X17000189
Financial support
G.P.P.L. thanks the Dirección General de Asuntos del Personal Académico (UNAM), and Consejo Nacional de Ciencia y Tecnología, for financial support during a sabbatical to New Zealand. G.P.P.L. also thanks the Programa de Apoyo a Proyectos de Investigación e Inovación Tecnológica (PAPIIT-UNAM) IN204514 for funding.
Conflict of interest
None.






