Psychosis refers to a subset of severe mental illness marked by delusions, hallucinations, and disorganized behavior. Schizophrenia is the prototypical psychotic disorder and it is defined by the presence of persistent psychotic symptoms and impaired functioning. However, psychosis manifests across a spectrum of conditions including major depressive disorder, bipolar disorder, schizoaffective disorder, delusional and schizophreniform disorder. Over 3% of individuals develop a psychotic disorder at some point during their lifetime but less than 1% are diagnosed with schizophrenia (Perala et al. Reference Perala, Suvisaari, Saarni, Kuoppasalmi, Isometsa and Pirkola2007). All forms of psychosis tend to emerge in early adulthood, cause persistent disability, and premature death (Smith, Reference Smith2011; Reininghaus et al. Reference Reininghaus, Dutta, Dazzan, Doody, Fearon and Lappin2015). Psychotic disorders are often preceded by early manifestations of psychopathology, including transitory psychotic symptoms that commonly occur in childhood (Kelleher et al. Reference Kelleher, Connor, Clarke, Devlin, Harley and Cannon2012). Childhood psychotic symptoms tend to be transient but they are associated with increased risk of psychotic disorders in adulthood (Poulton et al. Reference Poulton, Caspi, Moffitt, Cannon, Murray and Harrington2000; Fisher et al. Reference Fisher, Caspi, Poulton, Meier, Houts and Harrington2013) and may be considered antecedents to psychotic illness (Uher et al. Reference Uher, Cumby, Mackenzie, Morash-conway, Glover and Aylott2014). Knowledge of mechanisms involved in the etiology of psychosis may inform effective prevention. While a variety of risk factors have been identified, the causation of psychosis is still far from being understood. In this paper, we take stock of genetic and environmental factors associated with psychosis. Since there is substantial overlap of etiological factors across all types of severe mental illness (Uher & Zwicker, Reference Uher and Zwicker2017), we include evidence on the entire spectrum of psychotic disorders wherever possible. However, the largest body of data has been collected specifically on schizophrenia. We will review genetic and environmental contributions to psychosis separately before examining the joint effects of genetics and the environment. We will conclude by providing suggestions to guide future research.
The role of genetics in the development of psychosis
Psychosis runs in families. The strongest known predictor of risk is having a close biological relative who is affected (Gottesman et al. Reference Gottesman, Laursen, Bertelsen and Mortensen2010). Results from adoption and twin studies suggest that the familial clustering of psychosis is due largely to genetic factors. Among individuals with schizophrenia who were adopted at birth, increased rates of schizophrenia were found among their biological relatives and not within their adoptive families (Kety et al. Reference Kety, Wender, Jacobsen, Ingraham, Jansson and Faber1994). Additionally, twin studies show that monozygotic twins, who are genetically identical, are substantially more similar in their propensity to develop schizophrenia than are dizygotic twins, who share only half of their genetic material. The heritability of schizophrenia derived from twin studies is estimated to be 92% (Polderman et al. Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven and Visscher2015). Twin and family studies also suggest a substantial genetic contribution to psychotic symptoms in childhood (Zavos et al. Reference Zavos, Freeman, Haworth, McGuire, Plomin and Cardno2014). Taken together, this information strongly suggests that genetic factors significantly contribute to psychosis.
The last two decades have seen major progress in the identification of specific genetic variants that contribute to the risk of psychosis. To date, most molecular genetic investigations have focused on schizophrenia. Schizophrenia is a complex disorder and no single gene or genetic variant has been implicated as a necessary and sufficient causal factor. In linkage analysis, multiple loci across the genome have met the threshold for genome-wide or suggestive significance for an association with schizophrenia (Ng et al. Reference Ng, Levinson, Faraone, Suarez, DeLisi and Arinami2009). Due to the cost and difficulty associated with comprehensive genotyping, early investigations into specific molecular genetic contributions to psychosis focused on identifying associations between schizophrenia and a small number of genes thought to play a role in leading etiological hypotheses of psychosis. Using this hypothesis-driven method, associations were identified between several of these biologically plausible genes and schizophrenia. However initial reports often failed to replicate (Farrell et al. Reference Farrell, Werge, Sklar, Owen, Ophoff and O'Donovan2015). The candidate gene era culminated in the finding that associations between single nucleotide polymorphisms (SNPs) in top candidate genes previously reported to be associated with schizophrenia were consistent with chance expectation in the largest sample available at the time (Sanders et al. Reference Sanders, Duan, Levinson, Shi, He and Hou2008).
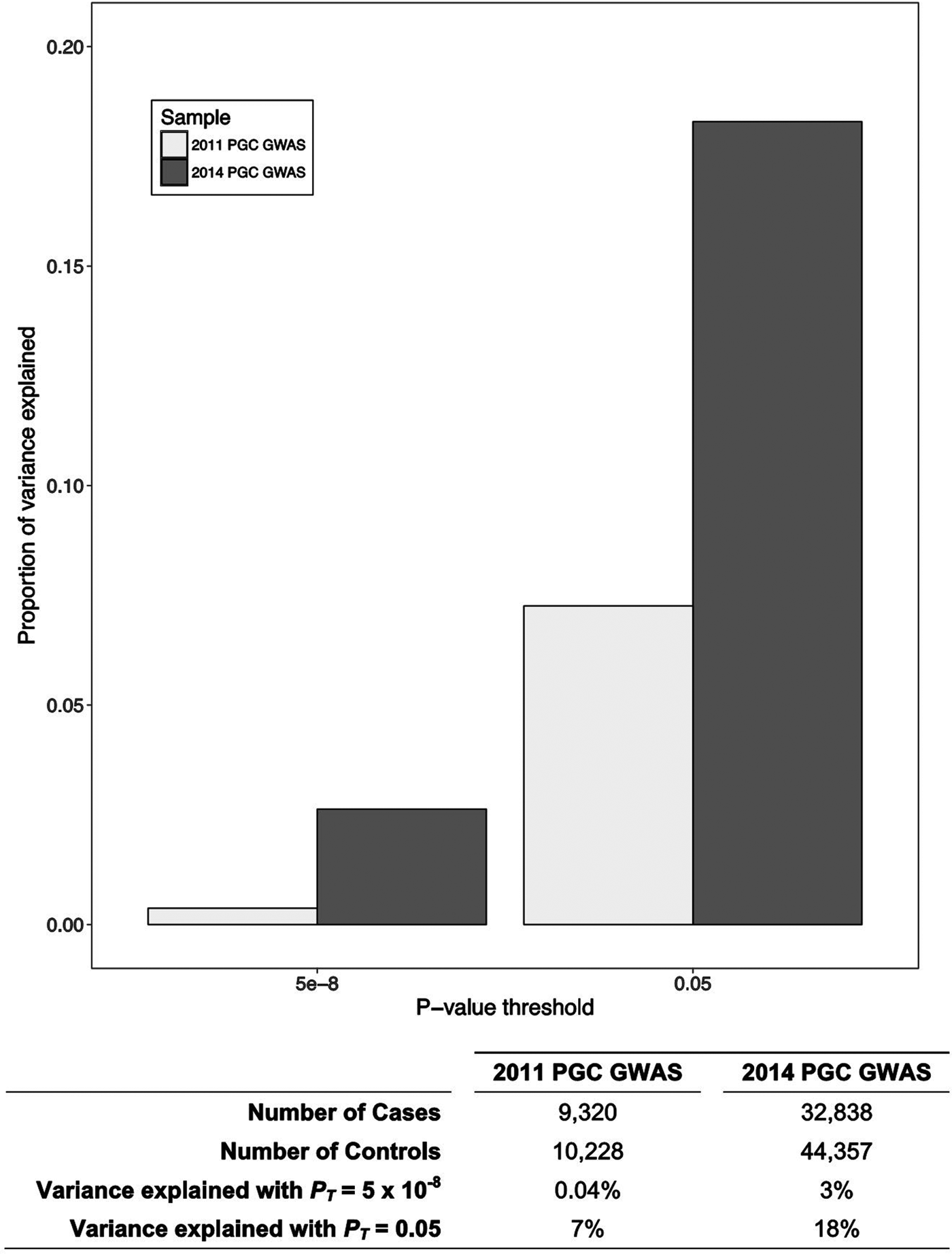
In the past decade, technological advances have enabled large-scale genomic studies and consortia have formed that have brought together sample sizes necessary for adequately powered genome-wide investigations (Sullivan et al. Reference Sullivan, Agrawal, Bulik, Andreassen, Borglum and Breen2017). The focus has shifted from candidate gene studies to the exploration of molecular genetic contributions to psychosis on a genome-wide scale. A case-control genome-wide association study (GWAS) conducted by the Schizophrenia Working Group of the Psychiatric Genomics Consortium identified over 100 independent loci that are associated with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). It is likely that substantially more (thousands) common variants contribute to the genetic liability for psychosis, because the predictive ability of polygenic risk scores (see Table 1 for definition) for schizophrenia improves with the inclusion of weakly associated variants in addition to variants significantly associated with the disorder (see Fig. 1). Some of the variants identified by GWAS fall within predictable genes (e.g. DRD2 – which encodes the dopamine receptor D 2, the target of most effective antipsychotic medications). However, the strongest association stemmed from variation within the major histocompatibility complex locus on chromosome 6, which is an area of the genome known for its role in immune function. This association is partly due to the many alleles of the complement component 4 (C4) genes (Sekar et al. Reference Sekar, Bialas, de Rivera, Davis, Hammond and Kamitaki2016). These alleles produce varying levels of C4A/C4B expression in the brain and alleles producing greater C4A expression were more associated with increased risk of schizophrenia. C4 is expressed in neurons and has been shown to mediate synaptic pruning during brain development in mice (Sekar et al. Reference Sekar, Bialas, de Rivera, Davis, Hammond and Kamitaki2016). These results implicate the immune system and complement activity in the causation of schizophrenia.
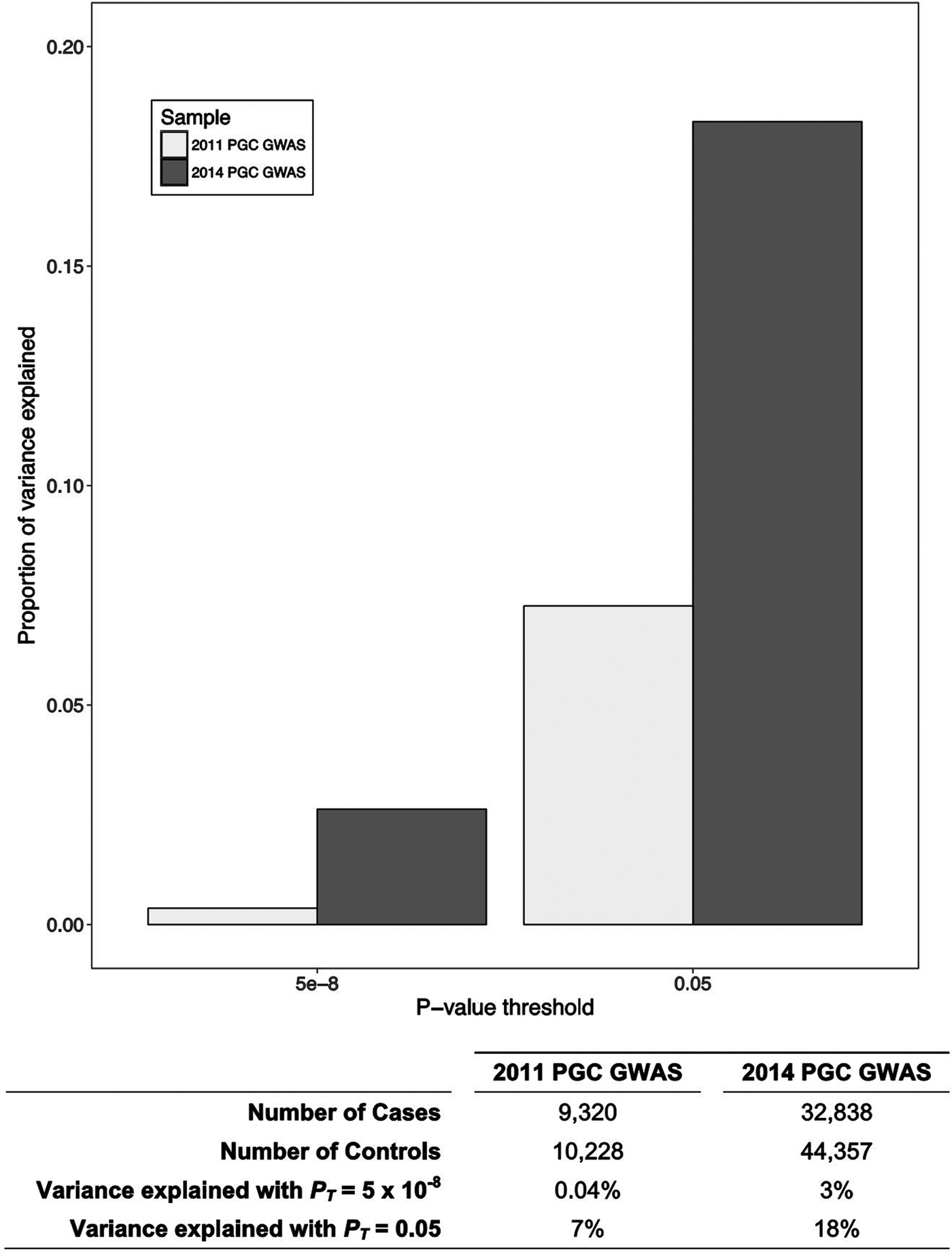
Fig. 1. The polygenic risk for schizophrenia. The proportion of variance explained by polygenic risk scores for schizophrenia produced using the Psychiatric Genomics Consortium (PGC) sample from 2011 (The Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium 2011) and 2014 (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) as the discovery samples, with p value thresholds (PT) at 5 × 10−8 (genome-wide significance) and 0.05. The proportion of variance explained increases with both sample size and with the inclusion of more, non-significantly associated variants. This suggests that schizophrenia is highly polygenic, and many more genetic loci contribute to disease risk than have been captured by the most recent GWAS.
Table 1. Terminology of gene–environment interplay

The results of genome-wide investigations of common SNPs suggest that genetic liability to schizophrenia arises from the accumulation of small effects of hundreds to thousands of variants across the genome. In addition to the cumulative effect of common SNPs, rare copy number variants and disruptive mutations are also enriched among individuals with schizophrenia compared with controls (Purcell et al. Reference Purcell, Moran, Fromer, Ruderfer, Solovieff and Roussos2014; Marshall et al. Reference Marshall, Howrigan, Merico, Thiruvahindrapuram, Wu and Greer2016). A specific deletion on chromosome 22, leading to 22q11.2 deletion syndrome, is associated with increased risk of psychosis. Approximately one in four individuals with this deletion will develop schizophrenia (Owen & Doherty, Reference Owen and Doherty2016). However, 22q11.2 deletion syndrome is not specific to schizophrenia, and is associated with a range of medical and psychiatric phenotypes (Niarchou et al. Reference Niarchou, Zammit, Van Goozen, Thapar, Tierling and Owen2014; Owen & Doherty, Reference Owen and Doherty2016). More recently, exome sequencing has enabled identification of rare variants that may alter protein function and are associated with schizophrenia (Girard et al. Reference Girard, Dion, Bourassa, Geoffroy, Lachance-Touchette and Barhdadi2015; Genovese et al. Reference Genovese, Fromer, Stahl, Ruderfer, Chambert and Landén2016; Singh et al. Reference Singh, Kurki, Curtis, Purcell, Crooks and McRae2016). Psychosis-associated rare variants, including deletions/duplications and copy number variants, are more prevalent in genes involved in neurodevelopment, e.g. neuregulin (Walsh et al. Reference Walsh, McClellan, McCarthy, Addington, Pierce and Cooper2008). Additionally, rare copy number variants have been identified in individuals with psychosis from densely affected families (Van Den Bossche et al. Reference Van Den Bossche, Strazisar, Cammaerts, Liekens, Vandeweyer and Depreeuw2013). Rare variants directly affecting protein function could individually have larger effects on psychosis risk than the common variants detected by genome-wide SNP analysis. The evidence suggests that genetic risk of psychosis is likely due to a combination of rare and common genetic variation.
The ability to identify and quantify genetic risk for psychosis allows us to investigate how genetic liability for illness manifests before disease onset. Genetic liability for schizophrenia is enriched among individuals with a family history (Bigdeli et al. Reference Bigdeli, Ripke, Bacanu, Lee, Wray and Gejman2016). The genetic burden of both rare and common variants is higher among individuals with early-onset compared with later-onset schizophrenia (Walsh et al. Reference Walsh, McClellan, McCarthy, Addington, Pierce and Cooper2008; Ahn et al. Reference Ahn, Gotay, Andersen, Anvari, Gochman and Lee2014, Reference Ahn, An, Shugart and Rapoport2016). In a general population sample, genetic risk for schizophrenia is associated with anxiety, negative symptoms, and worse neurodevelopmental outcomes (e.g. worse language fluency) in childhood and adolescence but, surprisingly, not with adolescent psychotic symptoms (Jones et al. Reference Jones, Stergiakouli, Tansey, Hubbard, Heron and Cannon2016; Riglin et al. Reference Riglin, Collishaw, Richards, Thapar, Maughan and O'Donovan2016). However, these findings warrant further investigation due to the high and non-random attrition of participants in this particular study (Jones et al. Reference Jones, Stergiakouli, Tansey, Hubbard, Heron and Cannon2016; Martin et al. Reference Martin, Tilling, Hubbard, Stergiakouli, Thapar and Davey Smith2016; Taylor et al. Reference Taylor, Jones, Sallis, Eusden, Stergiakouli and Davies2017). To find out when in development genetic risk for psychosis first manifests, it is necessary to establish large genotyped cohorts with high retention rates over longitudinal follow-ups with comprehensive phenotyping, including putative developmental precursors of psychotic illness.
Polygenic analyses across multiple disorders suggest that the genetic loci contributing to schizophrenia risk are also implicated in other mental and physical illnesses. There is substantial overlap between genetic liability to schizophrenia and bipolar disorder, major depressive disorder, and autism (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013b). To a smaller extent, schizophrenia-associated genetic variants also confer increased risk of inflammatory bowel disease, further supporting a role of the immune system in schizophrenia etiology (Pickrell et al. Reference Pickrell, Berisa, Liu, Ségurel, Tung and Hinds2016). In summary, the heritable contribution to psychosis is highly polygenic, arises from a combination of rare and common variation, and overlaps with other forms of pathology.
Environmental factors contributing to psychosis
The relative contributions of environmental and genetic factors to psychosis have been debated over time. Twin studies seem to suggest that less than 20% of the variance in liability to schizophrenia is explained by environmental influences unique to the individual and that environment shared between twins plays no role in the causation of schizophrenia (Sullivan et al. Reference Sullivan, Kendler and Neale2003; Polderman et al. Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven and Visscher2015). However, other typed of research studies strongly suggest a relatively smaller contribution of genetics and greater role of environment. Epidemiological studies show strong and consistent associations between multiple environmental exposures and psychosis (Marconi et al. Reference Marconi, Di Forti, Lewis, Murray and Vassos2016; Varese et al. Reference Varese, Smeets, Drukker, Lieverse, Lataster and Viechtbauer2012). Molecular genetic studies in unrelated individuals give substantially lower heritability estimates than twin studies (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013a; Sullivan et al. Reference Sullivan, Agrawal, Bulik, Andreassen, Borglum and Breen2017). In combination, the results of epidemiological research and the gap between twin and molecular heritability estimates suggest that environmental exposures likely play a more prominent role in the etiology of psychotic illness than was previously thought (Uher & Zwicker, Reference Uher and Zwicker2017).
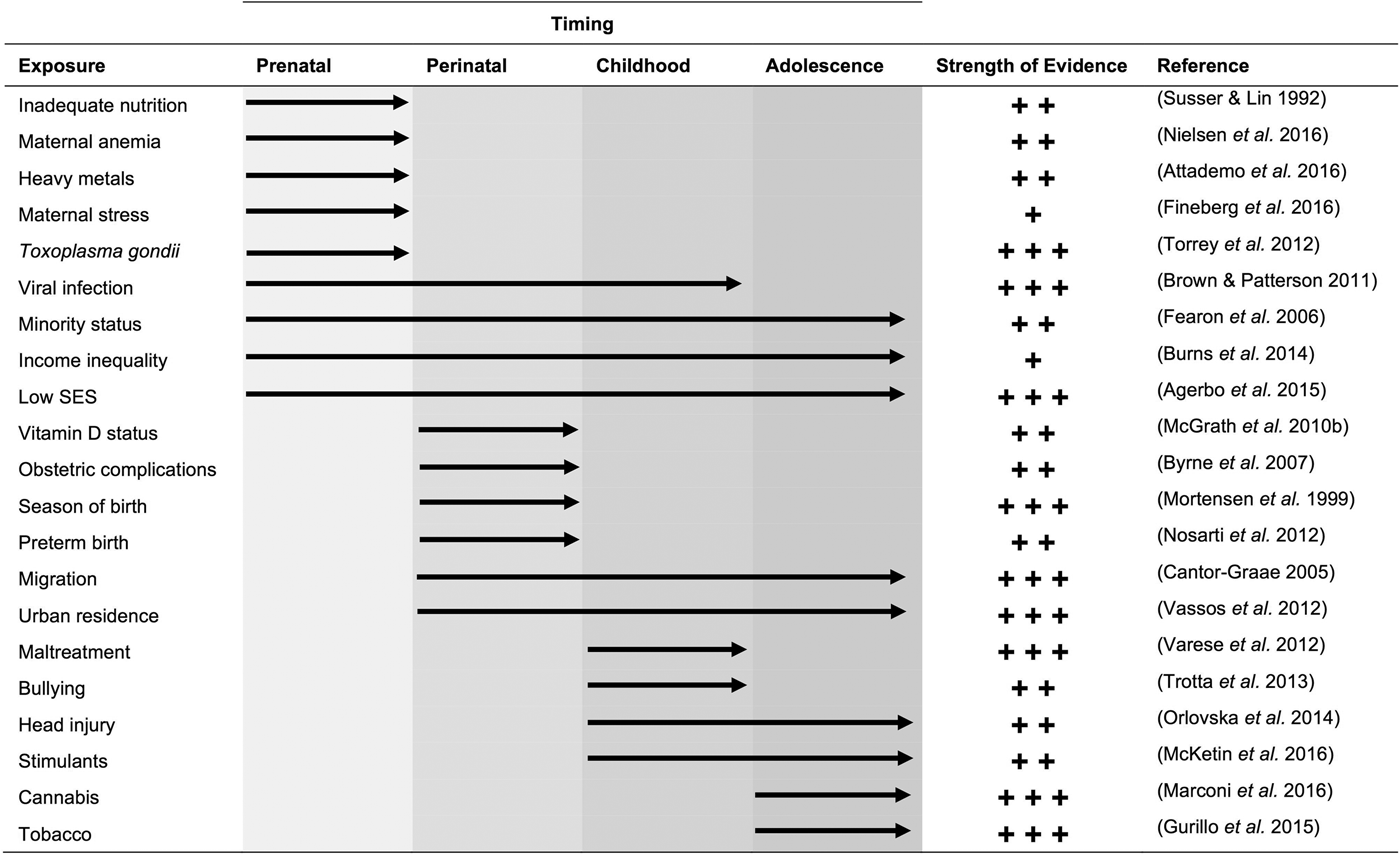
The major environmental exposures that have been implicated in psychosis are listed in Fig. 2. They can be clustered based on the stage in development when exposure could influence risk. Complex factors, such as parental socioeconomic status (SES) and income inequality tend to remain constant throughout development and have wide-reaching implications on health across the life course (Adler et al. Reference Adler, Boyce, Chesney, Cohen, Folkman and Kahn1994; Kennedy et al. Reference Kennedy, Kawachi, Glass and Prothrow-Stith1998). Other factors, including maternal viral infection, influence psychosis risk if individuals are exposed during a ‘sensitive’ period in development (Brown & Patterson, Reference Brown and Patterson2011). Most individuals, with and without psychotic illness, are exposed to at least one risk factor, which complicates the investigation of the roles of individual environmental exposures (Van Nierop et al. Reference Van Nierop, Janssens, Bruggeman, Cahn, De Haan and Kahn2013; Stepniak et al. Reference Stepniak, Papiol, Hammer, Ramin, Everts and Hennig2014).
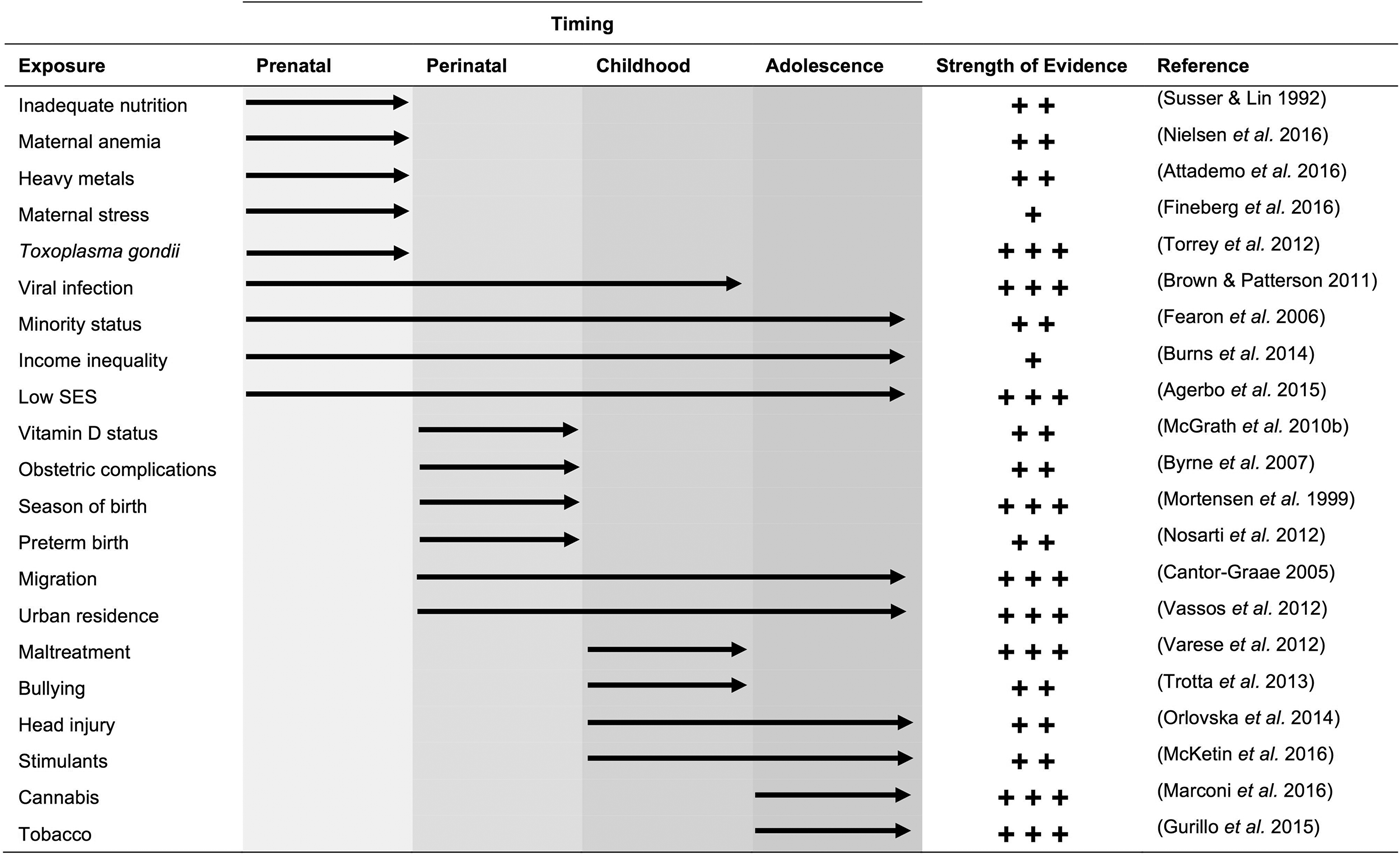
Fig. 2. Environmental factors associated with psychosis. The number of plus signs denotes the strength of evidence for the association: +++ indicates consistent evidence from multiple large-scale studies or a meta-analysis; ++ indicates evidence from multiple smaller studies or a strong association in a high-quality study; + indicates evidence from a single study, multiple small/low-quality studies, or few studies with conflicting reports. The reference provided for each exposure reflects a meta-analysis (where available) or the largest study available. The list is limited to environmental exposures and excludes risk factors that reflect conditions of the individual (e.g. inflammation).
In terms of pre- and perinatal risk factors, exposures that influence immune function have been linked to the development of psychotic illness (Mednick et al. Reference Mednick, Machon, Huttunen and Bonett1988; van Os & Selten, Reference van Os and Selten1998; Brown et al. Reference Brown, Schaefer, Wyatt, Goetz, Begg and Gorman2000, Reference Brown, Cohen, Harkavy-Friedman, Babulas, Malaspina and Gorman2001; Buka et al. Reference Buka, Cannon, Torrey and Yolken2008; Khashan et al. Reference Khashan, Abel, Mcnamee, Pedersen, Webb and Baker2008; Malaspina et al. Reference Malaspina, Corcoran, Kleinhaus, Perrin, Fennig and Nahon2008; Torrey et al. Reference Torrey, Bartko and Yolken2012; Fineberg et al. Reference Fineberg, Ellman, Schaefer, Maxwell, Shen and Chaudhury2016). It has been suggested that immune activation and subsequent inflammation could mediate the effects of pre- and perinatal insults, such as stress or infection, on psychosis risk by contributing to abnormal neurodevelopment (Deverman & Patterson, Reference Deverman and Patterson2009; Holloway et al. Reference Holloway, Moreno, Umali, Rayannavar, Hodes and Russo2013; Miller et al. Reference Miller, Culpepper, Rapaport and Buckley2013; Ménard et al. Reference Ménard, Pfau, Hodes and Russo2017). Individuals exposed to high levels of inflammation in utero are at increased risk of developing schizophrenia in adulthood (Canetta & Sourander, Reference Canetta and Sourander2014). Conversely, elevated levels of anti-inflammatory molecules in utero lower the risk of developing psychosis in adulthood (Allswede et al. Reference Allswede, Buka, Yolken, Torrey and Cannon2016). Results from animal studies suggest that the unfavorable effects of prenatal immune activation may extend across multiple generations. For example, using a mouse model, some pathological traits (e.g. reduced sociability) resulting from in utero exposure to immune activation were observable for up to three generations (Weber-Stadlbauer et al. Reference Weber-Stadlbauer, Richetto, Labouesse, Bohacek, Mansuy and Meyer2017). This suggests that epigenetic mechanisms or learned behavioral transmission may influence these traits.
Early adversities occurring in childhood, including physical, sexual, and emotional abuse, neglect and exposure to violence have been strongly implicated in the development of psychotic illness (Varese et al. Reference Varese, Smeets, Drukker, Lieverse, Lataster and Viechtbauer2012). More recently, involvement in bullying, both as a victim and as a bully, has been recognized as a contributor to both psychotic illness in adulthood and subclinical psychotic symptoms in adolescence (Schreier et al. Reference Schreier, Wolke, Thomas, Horwood, Hollis and Gunnell2009; Trotta et al. Reference Trotta, Di Forti, Mondelli, Dazzan, Pariante and David2013; Wolke et al. Reference Wolke, Lereya, Fisher, Lewis and Zammit2014). Childhood trauma is associated with increased levels of inflammatory markers in adulthood, which provides a possible mechanism through which childhood adversities could impact the development of psychosis (Baumeister et al. Reference Baumeister, Akhtar, Ciufolini, Pariante and Mondelli2016).
Exposures associated with psychosis that occur later in development are largely substance use-related. Abuse of psychostimulants is typically associated with acute psychosis, however, individuals with a family history of mental illness who use stimulants recreationally appear to be more vulnerable to persistent psychotic symptoms (Curran et al. Reference Curran, Byrappa and Mcbride2004; Li et al. Reference Li, Lu, Xiao, Li, He and Mei2014; Hajebi et al. Reference Hajebi, Amini, Kashani and Sharifi2016; McKetin et al. Reference McKetin, Gardner, Baker, Dawe, Ali and Voce2016). Interestingly, the link between stimulants and psychosis extends to children with a family history of mental illness taking stimulants to treat attention-deficit/hyperactivity disorder (MacKenzie et al. Reference MacKenzie, Abidi, Fisher, Propper, Bagnell and Morash-Conway2016). The risk of experiencing psychotic symptoms is more than four times higher among children taking prescribed stimulant medication compared with those who have never taken stimulants (MacKenzie et al. Reference MacKenzie, Abidi, Fisher, Propper, Bagnell and Morash-Conway2016). Regardless of family history status, cannabis use has been strongly and consistently associated with psychotic disorders (Moore et al. Reference Moore, Zammit, Lingford-hughes, Barnes, Jones and Burke2007; Gage et al. Reference Gage, Hickman and Zammit2015). In the case of cannabis exposure, many criteria of causality are met including a positive dose–response relationship between cannabis use and psychotic outcomes (Marconi et al. Reference Marconi, Di Forti, Lewis, Murray and Vassos2016), the temporal sequence of cannabis use preceding the onset of psychosis (Arseneault et al. Reference Arseneault, Cannon, Poulton, Murray, Caspi and Moffitt2002; Stefanis et al. Reference Stefanis, Dragovic, Power, Jablensky, Castle and Morgan2013; Kelley et al. Reference Kelley, Wan, Broussard, Crisafio, Cristofaro and Johnson2016), and consistent evidence for an association (Hill, Reference Hill1965). Additionally, a Mendelian randomization study (see Table 1) found that use of cannabis is causally associated with risk of schizophrenia and that cannabis users had a 37% increased risk of developing schizophrenia (Vaucher et al. Reference Vaucher, Keating, Lasserre, Gan, Lyall and Ward2017). There is also evidence that tobacco use could play a role in the development of psychosis (van Gastel et al. Reference van Gastel, MacCabe, Schubart, Vreeker, Tempelaar and Kahn2013; Gurillo et al. Reference Gurillo, Jauhar, Murray and MacCabe2015; McGrath et al. Reference McGrath, Alati, Clavarino, Williams, Bor and Najman2016). The fact that cannabis and tobacco are often used by the same individuals complicates the investigation into the nature of the individual relationships between cannabis and tobacco use and psychosis (Gage et al. Reference Gage, Hickman, Heron, Munafò, Lewis and Macleod2014). This highlights the necessity of gathering information on exposure to multiple factors when examining environmental contributors to psychosis.
Broad characteristics of the physical environment during development have also been associated with psychotic illness. Upbringing in an urban center is associated with increased risk of psychosis compared with rural upbringing (Vassos et al. Reference Vassos, Pedersen, Murray, Collier and Lewis2012). Risk increases with total time spent living in an urban setting (Pedersen & Mortensen, Reference Pedersen and Mortensen2001). This finding is not exclusive to adult illness; psychotic symptoms among children and adolescents are more frequent and more likely to progress to a first episode of psychosis among youth living in an urban environment (Polanczyk et al. Reference Polanczyk, Moffitt, Arseneault, Cannon, Ambler and Keefe2010; Dragt et al. Reference Dragt, Nieman, Veltman, Becker, van de Fliert and de Haan2011). The exact components of the urban environment that contribute to psychosis are not well-defined. Air pollution and toxins, such as heavy metals, are not likely to play a substantial role (Attademo et al. Reference Attademo, Bernardini, Garinella and Compton2017). A deficiency or excess of Vitamin D, which is produced in the skin when exposed to sunlight and may play a role in the regulation of gene expression, could explain a proportion of psychosis (McGrath, Reference McGrath1999; McGrath et al. Reference McGrath, Saari, Hakko, Jokelainen, Jones and Järvelin2004, Reference McGrath, Burne, Féron, MacKay-Sim and Eyles2010a, Reference McGrath, Eyles, Pedersen, Anderson, Ko and Burne2010b). It has been suggested that vitamin D deficiency is more prevalent in urban centers, possibly due to less sun exposure (Holick, Reference Holick1995). In the case of childhood and adolescent psychotic symptoms, neighborhood factors including low social cohesion and high crime rate partially explain the elevated risk for psychotic symptoms among children living in urban areas (Polanczyk et al. Reference Polanczyk, Moffitt, Arseneault, Cannon, Ambler and Keefe2010; Newbury et al. Reference Newbury, Arseneault, Caspi, Moffitt, Odgers and Fisher2016).
Although some environmental risk factors for psychosis have been identified, it is difficult to separate the effects of individual factors because exposures often cluster within the same person (Gage et al. Reference Gage, Hickman, Heron, Munafò, Lewis and Macleod2014; Sideli et al. Reference Sideli, Fisher, Murray, Sallis, Russo and Stilo2015). Similar to the notion of many genetic factors of small effect sizes contributing to the genetic risk of psychosis, it has been suggested that environmental risk is also attributable to the cumulative contribution of many exposures (Van Nierop et al. Reference Van Nierop, Janssens, Bruggeman, Cahn, De Haan and Kahn2013). Exposure to a greater number of environmental risk factors was associated with earlier age at onset of psychotic symptoms and first-episode of psychosis (Stepniak et al. Reference Stepniak, Papiol, Hammer, Ramin, Everts and Hennig2014; O'Donoghue et al. Reference O'Donoghue, Lyne, Madigan, Lane, Turner and O'Callaghan2015). An aggregate ‘polyenviromic’ score predicted the onset of psychosis among youth with a family history of psychotic illness (Padmanabhan et al. Reference Padmanabhan, Shah, Tandon and Keshavan2017). Although exposure to certain environments increases the risk of psychosis, only a minority of individuals exposed to these factors will become ill. Genetic differences may render some individuals more vulnerable or resilient to the impact of environmental exposures.
Gene–environment interplay
Psychotic illness in a given individual arises due to a combination of genetics and environment. The term gene–environment interplay captures the combined contributory effects of both genetics and environment to psychosis. Gene–environment interplay encompasses gene-environment correlation (rGE) and gene–environment interaction (GxE; see Table 1 for definitions).
Gene-environment correlation
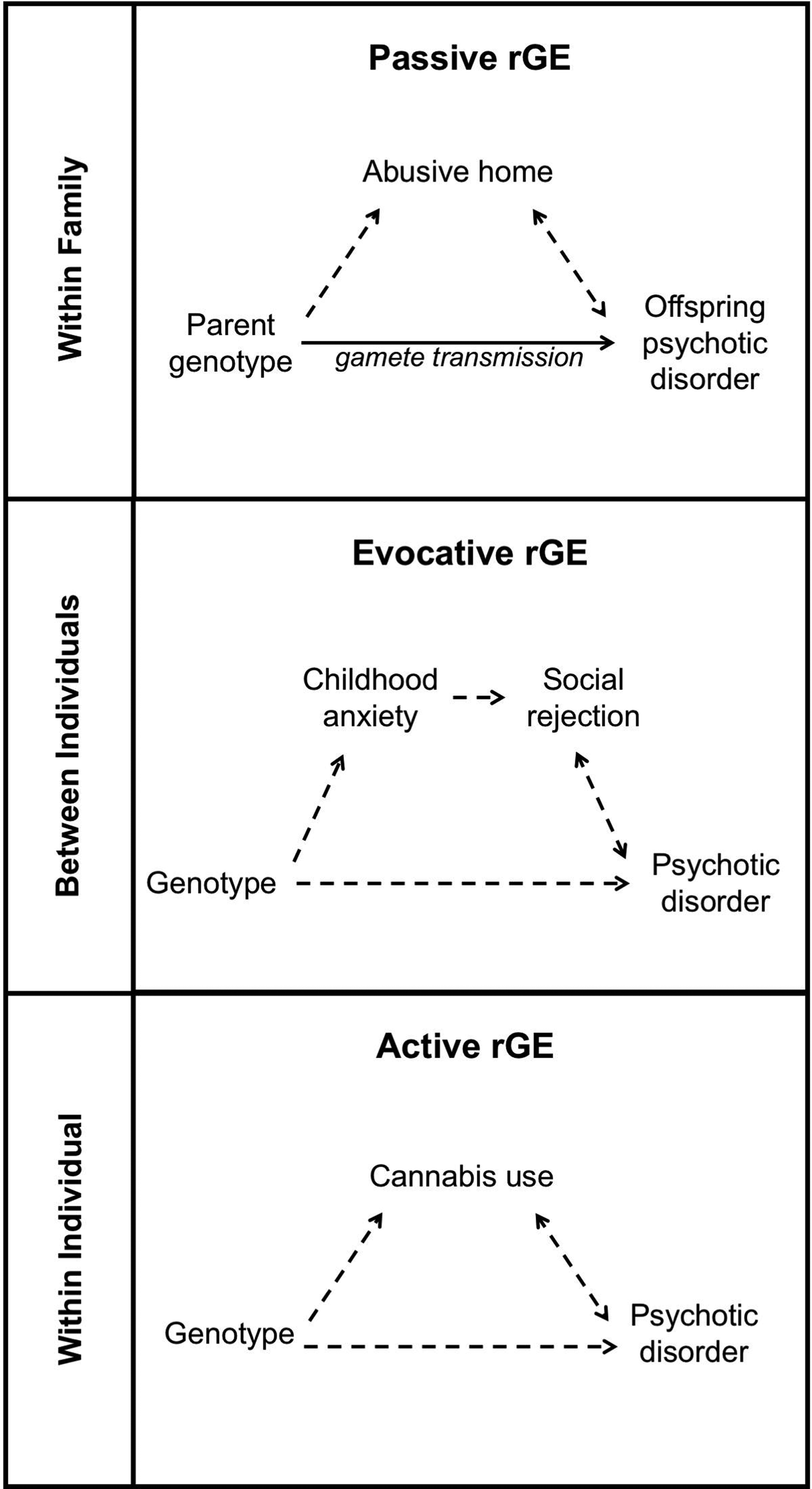
Exposure to specific environments is not random. Gene-environment correlation (rGE) refers to this non-random relationship between genotype and exposure. It is important to assess the possible contribution of rGE when investigating causal contributors to psychosis. Three forms of rGE have been widely described: passive rGE, evocative (or reactive) rGE and active rGE (Plomin et al. Reference Plomin, DeFries and Loehlin1977), see Fig. 3.
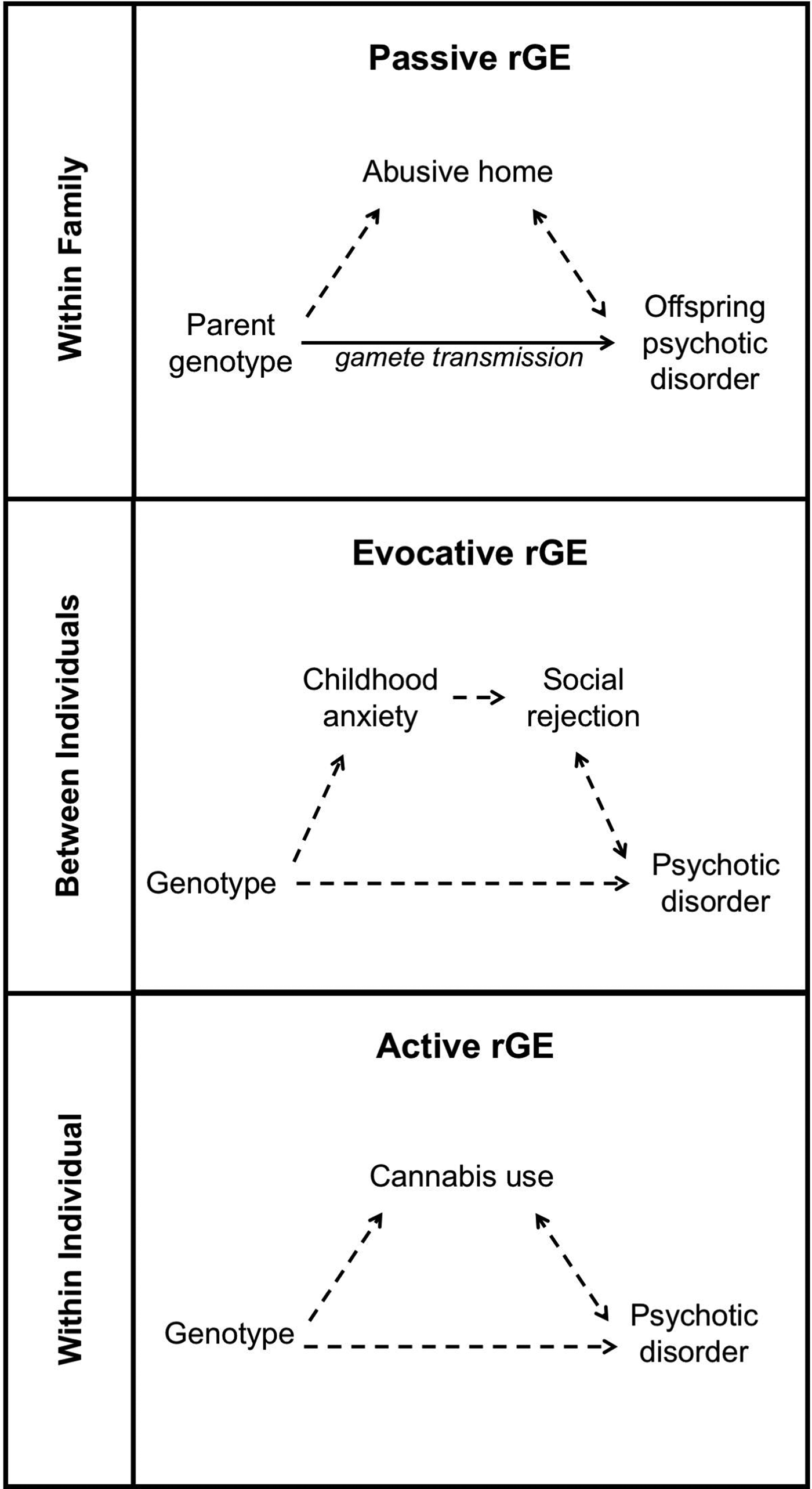
Fig. 3. Gene-environment correlations and psychosis. The three panels illustrate passive, evocative and active gene-environment correlations (rGE), base on specific examples that are relevant to the etiology of psychosis. The same examples are described and referenced in the text section Gene-Environment Correlation.
Passive gene-environment correlation
Passive rGE describes the association between genotype and rearing environment, both of which are influenced by an individual's parents’ genes. For example, the association between childhood abuse and genetic risk for psychosis represents a potential passive rGE. Children of parents with psychosis are at increased risk of experiencing childhood maltreatment and individuals who experience abuse are more likely to develop psychosis (Walsh et al. Reference Walsh, MacMillan and Jamieson2002; Fisher et al. Reference Fisher, McGuffin, Boydell, Fearon, Craig and Dazzan2014). However, it is unclear whether the association between childhood maltreatment and psychosis arises because abuse directly causes psychosis or because genetic factors increase both the likelihood of experiencing abuse and the propensity to develop psychosis. If the latter is true, the association between childhood abuse and psychotic illness represents a passive gene-environment correlation. Adoption studies provide a useful design to tease apart passive rGE from G × E. Using this methodology, it has been possible to demonstrate that individuals with a family history of schizophrenia are truly more vulnerable to the psychosis-inducing effects of childhood adversity than are individuals without a family history (Tienari et al. Reference Tienari, Ynne, Sorri and Lahti2004).
Evocative gene-environment correlation
Personal characteristics determine how individuals interact in social situations, and as a result, influence the responses they will elicit from others. Evocative rGE describes how differences in genotype will evoke different reactions. As an example, genetic risk of schizophrenia is associated with the presence of childhood anxiety disorders (Jones et al. Reference Jones, Stergiakouli, Tansey, Hubbard, Heron and Cannon2016). Anxious children may evoke a different response from peers than their less anxious counterparts, resulting in an evocative rGE. Thus, anxious children may face rejection in social interactions with peers, which could, in turn, increase their risk of developing psychosis (Collishaw et al. Reference Collishaw, Pickles, Messer, Rutter, Shearer and Maughan2007).
Active gene-environment correlation
Individual behaviors and preferences are also partially determined by genetics. By influencing these factors, genotype also influences the experiences that people will seek out. Active rGE describes how genetic variation can contribute to differences in the likelihood of exposure to environments. For example, individuals at high genetic risk for schizophrenia are more likely to use cannabis (Verweij et al. Reference Verweij, Abdellaoui, Nivard, Sainz Cort, Ligthart and Draisma2017). Genotype, therefore, influences both the propensity to develop psychosis and the likelihood of being exposed to cannabis. Active rGE could account for a portion of the association between cannabis use and psychosis. However, genetic risk for schizophrenia only explains a small proportion (0.5%) of the variance in cannabis use (Verweij et al. Reference Verweij, Abdellaoui, Nivard, Sainz Cort, Ligthart and Draisma2017). Therefore, it is unlikely that rGE fully explains the association between cannabis use and psychosis. It does, however, bring attention to the need to consider rGE before making causal interpretations that assume relative independence of genetic and environmental factors.
Gene–environment interaction
Exposure to certain environmental factors increases the risk of psychosis, however, these adverse environments do not affect everyone equally. Some individuals remain healthy even when exposed to multiple known risk factors whereas others will go on to develop a psychotic disorder (Collishaw et al. Reference Collishaw, Pickles, Messer, Rutter, Shearer and Maughan2007; Tottenham, Reference Tottenham2013). Genetic factors may render certain individuals more vulnerable to the impact of environmental exposures, resulting in a gene–environment interaction. Two sets of findings suggest that G × E plays a substantial role in the development of psychosis. First, much larger heritability estimates of schizophrenia have been obtained from twin studies than from molecular genetic studies using unrelated individuals (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013a). Second, despite the fact that epidemiological studies have robustly demonstrated that risk factors shared within families (such as minority status, urbanicity, or low SES) are among the top environmental contributors to psychosis, twin studies suggest that shared environment plays a very minor or no role (Polderman et al. Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven and Visscher2015). The best estimate of shared environment contribution across twin studies is actually a negative number (Polderman et al. Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven and Visscher2015). These implausible findings can be explained by the manner in which heritability is calculated in twin studies: the interplay between genetic factors and environmental variables shared within a family are attributed to genetics and inflate heritability estimates while reducing the estimated contribution of shared environment (Taylor, Reference Taylor2007; Uher & Zwicker, Reference Uher and Zwicker2017). Therefore, G × E offers a plausible explanation for these discrepant findings.
In addition to providing an explanation for the conflicting heritability estimates from twin studies, molecular genetic studies and epidemiology, identification of G × E could offer additional insight into both genetic and environmental contributions to psychosis. First, G × E research can lead to the identification of novel genetic contributors to psychosis that may not otherwise be identified in case-control studies (Børglum et al. Reference Børglum, Demontis, Grove, Pallesen, Hollegaard and Pedersen2013). Additionally, identification of G × E is valuable because unlike genetics, environment is malleable and can be modified selectively among those at high genetic risk. Therefore, identifying G × E could provide the opportunity for targeted interventions to minimize or eliminate exposure to environmental contributors to psychosis, particularly exposures that are under the control of the individual, such as cannabis, tobacco and stimulant use. Despite the significant role of G × E in psychosis and the potential value of G × E knowledge, identification of G × Es to date has been slow.
Gene–environment interaction by proxy
Initial investigation of G × E in psychosis relied on the family history of severe mental illness as a proxy fo genetic risk. Using this method, interactions between ‘genetic liability’ for psychotic illness and exposure to environmental factors have been identified. Individuals with a family history of psychotic illness appear to be particularly sensitive to the effects of multiple environmental contributors to psychosis including cannabis use (GROUP investigators, 2011; van Winkel & GROUP investigators, Reference van Winkel2015), urban upbringing (Van Os et al. Reference Van Os, Hanssen, Bak, Bijl and Vollebergh2003, Reference Van Os, Pedersen and Mortensen2004), and maternal infection (Clarke et al. Reference Clarke, Tanskanen, Huttunen, Whittaker and Cannon2009). There have also been reports in which no interaction was found between family history and known environmental risk factors, such as childhood maltreatment (Fisher et al. Reference Fisher, McGuffin, Boydell, Fearon, Craig and Dazzan2014; Trotta et al. Reference Trotta, Di Forti, Iyegbe, Green, Dazzan and Mondelli2015). Other factors, such as maternal depression during pregnancy, selectively increases the risk of psychosis among those with at least one parent affected by psychotic illness (Mäki et al. Reference Mäki, Riekki, Miettunen, Isohanni, Jones and Murray2010). However, since there is overlap in the genetic factors contributing to depression and psychosis, this result could also reflect a gene-environment correlation or interaction between multiple genetic factors (Han et al. Reference Han, Pouget, Slowikowski, Stahl, Lee and Diogo2016). Gene-environment studies using proxy measures are inherently limited because a family history of illness is not equivalent to the genetic contribution to psychosis and because genetic variants that increase sensitivity to the environment may be distinct from genetic variants that directly increase the risk of illness. The applicability of findings using proxy measures of genetic contribution to illness is also limited because the comprehensive family history of mental illness is not always known.
Gene–environment interactions involving molecular genetic variants
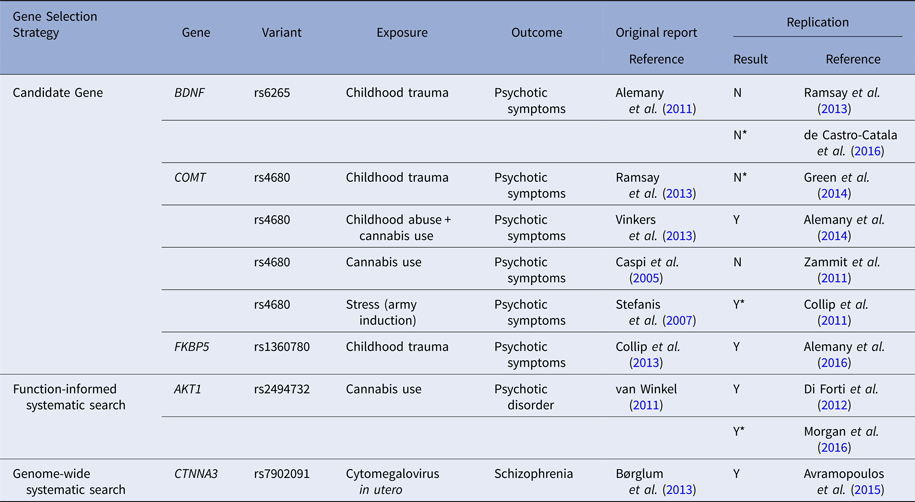
The search for gene–environment interactions involving specific molecular genetic variants began by testing the interaction between environmental risk factors for psychosis and variants within candidate genes. These studies faced the same challenges as candidate gene association studies discussed earlier. This approach relies on correctly selecting both the genetic variant and environmental exposure of interest based on prior knowledge. Surprisingly, this approach has led to the identification of interactions between variants in a handful of genes and environmental factors influencing the risk of psychosis (see Table 2). Replication of results, however, has been inconsistent. For example, carriers of the methionine-encoding allele of BDNF [Val66Met (rs6265)] have been shown to be at increased risk of experiencing psychotic symptoms after being exposed to childhood trauma (Alemany et al. Reference Alemany, Arias, Aguilera, Villa, Moya and Ibanez2011), but attempts to replicate this finding have either found no interaction between genotype and exposure to trauma (Ramsay et al. Reference Ramsay, Kelleher, Flannery, Clarke, Lynch and Harley2013), or found discordant results regarding which allele (valine-encoding v. methionine-encoding) increases psychosis risk following exposure (de Castro-Catala et al. Reference de Castro-Catala, van Nierop, Barrantes-Vidal, Cristóbal-Narváez, Sheinbaum and Kwapil2016). More systematic searches, involving screening hundreds of polymorphisms across functionally defined groups of genes, have been carried out in search of a G × E. This methodology led to the identification of an interaction between cannabis use and a SNP in AKT1, a gene encoding a serine/threonine kinase involved in the transduction of signal following cannabinoid receptor activation (van Winkel, Reference van Winkel2011). Homozygous carriers of the C allele at rs2494732 in AKT1 appear to be more vulnerable to the psychosis-inducing properties of cannabis. In a replication study, individuals with the C/C genotype at rs2494732 who used cannabis daily were found to be at 7-fold increased risk of developing psychotic illness compared with T allele homozygotes (Di Forti et al. Reference Di Forti, Iyegbe, Sallis, Kolliakou, Falcone and Paparelli2012). This finding has been replicated in two independent samples since the original report and therefore likely represents a true G × E (Di Forti et al. Reference Di Forti, Iyegbe, Sallis, Kolliakou, Falcone and Paparelli2012; Morgan et al. Reference Morgan, Freeman, Powell and Curran2016).
Table 2. Molecular gene–environment interactions in psychosis

The table lists specific molecular gene–environment interactions for which there is at least one published replication attempt. The first column classifies the G × Es based on the initial search strategy into G × Es identified based on focused candidate gene testing, systematic searches including polymorphisms that tag genetic variation in a functionally defined group of genes and genome-wide systematic search. For replication results, ‘Y’ denotes a replication, ‘Y*’ denotes a replication with a different but similar exposure or outcome from the original report, ‘N’ denotes a failed replication attempt, and ‘N*’ denotes a replication attempt where an interaction between the variant and the environment of interest was found but the sensitive allele differed from the original report.
Comprehensive genome-wide search strategies, referred to as genome–wide environment interaction studies (GWEIS), have also been conducted to systematically search for G × E. The first GWEIS in psychosis found a significant interaction in a genome-wide test examining the interaction between in utero exposure to cytomegalovirus (CMV) infection and hundreds of thousands of common SNPs across the genome (Børglum et al. Reference Børglum, Demontis, Grove, Pallesen, Hollegaard and Pedersen2013). This led to the identification of a single G × E between CMV infection and a variant within a gene (CTNNA3) not previously associated with psychosis (Børglum et al. Reference Børglum, Demontis, Grove, Pallesen, Hollegaard and Pedersen2013). Carriers of the minor allele at rs7902091 in CTNNA3 exposed to CMV in utero are at increased risk of developing schizophrenia compared with those who do not carry the minor allele and those who were not exposed to CMV. A genome-wide approach to G × E research is likely to be superior to hypothesis-driven approaches, as many schizophrenia-associated genetic loci that have been identified to date are found within genes that were not previously suspected to be implicated in psychosis (Collins et al. Reference Collins, Kim, Sklar, O'Donovan and Sullivan2012). Due to a large number of loci being tested, GWEIS require very large samples to be adequately powered – even when examining interactions between common variants and common exposures. Although it is likely that G × Es play a significant role in the development of psychosis, no individual G × E identified thus far explains a substantial proportion of cases. The potential gene–environment interactions involved in the early manifestation of psychosis in childhood and adolescence remain to be examined.
Gene–environment interactions and polygenic scores
It is possible to summarize the effects of hundreds to thousands of variants across the genome as a single polygenic score. Polygenic risk scores calculated based on the most recent case-control GWAS for schizophrenia have been used to test G × E across the genome. One study derived a score from a few thousand schizophrenia-associated genetic variants located within coding and regulatory regions and found that this score interacted with winter birth to increase the risk of developing schizophrenia (Hong Lee et al. Reference Hong Lee, Byrne, Hultman, Kahler, Vinkhuyzen and Ripke2015). Another study found no interaction between polygenic risk score for schizophrenia and childhood adversity (Trotta et al. Reference Trotta, Iyegbe, Di Forti, Sham, Campbell and Cherny2016). The authors of this study concluded that their results support a model whereby genetic and environmental factors contribute independently to psychosis risk.
Polygenic risk scores for schizophrenia may not be the ideal method to detect G × E because sensitivity to the environment and risk of illness may be determined by distinct subsets of genetic variants. It has been suggested that the individuals who are most severely impacted by adverse environments may also benefit most from positive ones. This is referred to as differential susceptibility (Belsky & Pluess, Reference Belsky and Pluess2009). The differential susceptibility hypothesis posits that the genetic variants that render individuals sensitive to environmental influences may be distinct from genetic factors that directly influence the risk of psychopathology (Belsky & Pluess, Reference Belsky and Pluess2009). Therefore, G × E involving variants contributing to differential susceptibility will not be captured when using a polygenic risk score for schizophrenia (Belsky & Pluess, Reference Belsky and Pluess2009). A ‘polygenic sensitivity score’ indexing overall sensitivity to the environment (both positive and negative) predicted both the effects of parenting on emotional problems and response to psychological treatment among children with anxiety disorders (Keers et al. Reference Keers, Coleman, Lester, Roberts, Breen and Thastum2016). This sensitivity score did not, however, predict psychopathology directly, thus providing evidence that environmental sensitivity is genetically distinct from illness (Keers et al. Reference Keers, Coleman, Lester, Roberts, Breen and Thastum2016). Like polygenic risk scores for schizophrenia, the predictive ability of the polygenic sensitivity score improved with the inclusion of a greater number of less significantly associated variants, suggesting that genetic contribution to environmental sensitivity is also dispersed over a large number of loci across the genome. A polygenic sensitivity score has yet to be tested in the etiology of psychosis or its early developmental antecedents.
Conclusions, gaps in evidence and future directions
The findings of the past decade have led to a better understanding of the etiology of psychosis but also highlight a need to shift the direction of future research. Advances in the availability of genome-wide technology have uncovered the massively polygenic nature of psychosis. Epidemiological investigations have identified many environmental exposures that are associated with psychosis, however, the effect of these exposures differs between individuals. It is likely that genetic factors influence the impact of environmental exposures, and that genetic sensitivity to the environment is also highly polygenic. Identification of specific gene–environment interactions has proven to be difficult and has been hindered by lack of environmental measurement in large genetic studies. GWEIS can detect multiple gene–environment interactions in a systematic manner, but this methodology is limited in terms of statistical power. Polygenic environmental sensitivity scores may facilitate future genome-wide investigation of G × E in moderately large samples.
Despite the progress of the past decade, there remain important gaps in knowledge. Although genotyping technologies have improved in efficiency and affordability, the commonly used genome-wide genotyping methods do not provide full coverage of the genome. Thus, despite their names, genomic methods such as GWAS, GWEIS, and polygenic risk scores -environment interaction studies still leave a proportion of genetic variation unexplored. Additionally, most investigations of the genetic contribution to psychosis have been performed by comparing the genotypes of individuals diagnosed with schizophrenia to controls who do not have schizophrenia. This approach, though a necessary first step, limits the generalizability of polygenic risk scores from schizophrenia to other forms of psychosis. Indeed, a polygenic risk score for schizophrenia may predict other psychotic disorders less accurately than narrowly defined schizophrenia (Vassos et al. Reference Vassos, Di Forti, Coleman, Iyegbe, Prata and Euesden2017). Additionally, most large-scale investigations of the genetic contribution to schizophrenia have relied on ethnically homogenous samples of white individuals of European descent. This limits the generalizability to other ethnicities. In a sample of individuals of African descent, the proportion of variance in psychosis case-control status explained by polygenic risk scores for schizophrenia is approximately 1/9th of the proportion of variance explained in a European sample (1.1% v. 9.4%; Vassos et al. Reference Vassos, Di Forti, Coleman, Iyegbe, Prata and Euesden2017). To improve the utility of genetic risk information, future gene-environment investigations should cover a broader range of psychosis phenotypes in ethnically diverse samples.
Samples with a thorough assessment of both psychopathology and environment at multiple points in development will likely provide the most valuable information on gene-environment causation. Psychosis typically emerges in the second decade of life and it is often preceded by different and milder forms of mental illness. It is therefore essential to examine environmental exposures and psychopathology earlier in life, prior to disease onset. These studies should begin as early as during pregnancy or infancy and continue longitudinally across development. Regular assessment across development will allow us to establish true age at onset and track the development of psychosis over time. Large cohorts with genome-wide SNP genotyping and/or whole genome sequencing and comprehensive assessment of exposures and psychopathology across development are needed for this investigation. Important steps towards establishing such cohorts are currently underway (Korver et al. Reference Korver, Quee, Boos, Simons and de Haan2012; Uher et al. Reference Uher, Cumby, Mackenzie, Morash-conway, Glover and Aylott2014; Thorup et al. Reference Thorup, Jepsen, Ellersgaard, Burton, Christiani and Hemager2015; Kooijman et al. Reference Kooijman, Kruithof, van Duijn, Duijts, Franco and van IJzendoorn2016). To expand and combine information from these cohorts, future work should focus on the formation of large-scale, transparent collaborations and data sharing mechanisms.
Acknowledgements
The work on this manuscript has been completed thanks to funding from the Canada Research Chairs Program (reference number 231397) and grants from the Canadian Institutes of Health Research (Grant reference numbers 124976, 142738 and 148394), Nova Scotia Health Research Foundation and the Dalhousie Medical Research Foundation awarded to Dr Rudolf Uher.