INTRODUCTION
Malaria represents an enormous global health problem, causing immense morbidity and mortality (defined here as virulence) worldwide (Snow et al. 2005). Malaria infection causes a wide spectrum of virulence, resulting in an estimated 2 million deaths each year (Alles, 1998). We are interested in understanding the causes of this variation in virulence. Both field and rodent studies have shown that while some virulence is directly linked to parasite load (Langhorne et al. 1998; Mackinnon and Read, 2004), some is immunopathological, arising as a result of the host's immune response to the parasite (Dodoo et al. 2002; Torre et al. 2002). The ‘cytokine theory’ of malaria, for example, hypothesizes that cytokines contribute to disease (Clark et al. 2004), and the potent pro-inflammatory cytokine tumour necrosis factor alpha (TNF-α) has been shown to have dual roles in malaria infection: inhibiting in vivo parasite growth and controlling infection on the one hand (Langhorne et al. 1989; Stevenson et al. 1995; Jacobs et al. 1996; Sam et al. 1999; Li and Langhorne, 2000), while causing immune-mediated disease on the other (Grau et al. 1987; Clark and Chaudhri, 1988a,b; Kern et al. 1992).
We are particularly interested in whether the host immune response contributes to the different virulence outcomes induced by genetically distinct P.c.c. infections. Studies using Plasmodium chabaudi chabaudi (P.c.c.), the immunologically best-characterized rodent model of malaria, have focused largely on a single clone: AS (Langhorne et al. 1989; Stevenson et al. 1995; Jacobs et al. 1996; Sam et al. 1999; Li and Langhorne, 2000). However, primary infections by other P.c.c. clones have been shown to induce a spectrum of virulence phenotypes in a given inbred mouse strain, supporting a role for parasite strain in virulence determination (Mackinnon and Read, 1999a,b; Ferguson et al. 2003). Strain-specific variation in malaria virulence during human P. falciparum infections has also been reported (Gravenor et al. 1995; Chotivanich et al. 2000). Because high serum TNF-α concentrations are associated with severe infection (Grau et al. 1989; Molyneux et al. 1991, 1993) and malaria parasites directly stimulate the production of TNF-α from macrophages (Bate et al. 1992; Bate et al. 1988, 1989, 1992; Bate and Kwiatkowski, 1994), we hypothesized that parasite genetic differences in induction of systemic TNF-α may contribute to clone differences in virulence. For example, distinct P.c.c. clones may achieve high virulence by (i) growing to high parasite densities in the host, (ii) inducing a large TNF-α response, or (iii) through a combination of these factors.
Ignoring the role parasite genotype plays in determining immune-mediated virulence may lead to conclusions which are parasite strain- or clone-specific. In the field of Leishmania, the role mouse strain played in disease outcome was the initial focus and led to conflicting data until it was recognized that parasite genotype could help to explain reported differences in disease outcome (Kebaier et al. 2001; Ritter et al. 2004). For example, infection with a range of L. major strains in BALB/c mice resulted in distinct immune responses and a spectrum of virulence, from asymptomatic to visceral leishmaniasis (Kebaier et al. 2001). In addition, the anaemia induced during rodent Trypanosoma infections (Naessens et al. 2005) and the outcome of toxoplasmosis in rodent models (Mordue et al. 2001) have been shown to depend on the immune response induced by the particular host-parasite combination. For malaria, numerous studies have shown that mouse strains vary in their ability to control and clear primary infections (Li et al. 2001). However, immunological analysis of the role of the parasite strain in determining the outcome of malaria has largely been overlooked, with the majority of rodent studies focusing on a single parasite strain or clone from a given Plasmodium species.
In the present study, we took two approaches to understand the role TNF-α plays in response to infection with distinct malaria genotypes. First, we investigated whether the level of virulence induced by the genetically distinct P.c.c. clones correlated with a specific host cytokine response. We found that more virulent clones induced a greater plasma TNF-α response. To directly test whether the induction of a large TNF-α response is the reason some clones are more virulent, we then blocked TNF-α signalling by administering a soluble form of the TNF Receptor 1 (TNFR-Ig) and followed in vivo virulence, parasite dynamics and cytokine responses during distinct P.c.c. infections. We found that blocking TNFR-1 signalling protected against weight loss, but the level of protection did not differ among clones.
MATERIALS AND METHODS
Mice and parasites
Plasmodium chabaudi chabaudi (P.c.c.) clones were originally derived from thicket rat isolates and cryopreserved (Walliker et al. 1971). Clones were stored as frozen stabilities, with subscript codes denoting their exact clonal history. In both experiments we used the following clones; CW640, AS11915, BC200 and AJ4762 (herein referred to as CW, AS, BC and AJ). These 4 genetically distinct P.c.c. clones were selected based on the range of virulence they induce in C57BL/6 female mice; clones BC and AJ are virulent and clones AS and CW substantially less virulent in terms of weight and red blood cell loss (Mackinnon and Read, 1999a). Clone differences in virulence are maintained across mouse strain (de Roode et al. 2005; Mackinnon et al. 2002). In both Experiments 1 and 2, hosts were 6 to 8-week-old female inbred C57BL/6 mice (Harlan, UK or bred at animal facilities of the University of Edinburgh, respectively). Mice were housed in filter-top cages, maintained in a 12 h[ratio ]12 h light-dark cycle, fed 41B maintenance diet (Harlan, UK) and their drinking water was supplemented with 0·05% para-amino benzoic acid (Jacobs, 1964).
Experimental design, parasite inoculation and in vivo antibody treatment
Infections were initiated with an i.p. injection of 0·1 ml of inoculum containing 106 red blood cells (RBCs) parasitized by one of the above 4 malaria clones. The inoculum was prepared from parasite donor mice through the dilution of infected blood in calf serum solution (50% Ringer's solution (27 mM KCL, 27 mM CaCl2, 0·15 M NaCl), 50% heat-inactivated calf-serum, 20 units heparin/ml mouse blood). Control mice received a 0·1 ml inoculum of 106 naïve RBCs and were subject to the same treatment regimes as experimental mice.
In Exp. 1, we investigated whether clone differences in malaria virulence could be explained by differences in plasma levels of TNF-α, interleukin [IL]-6, IL-12p70, IL-10 or interferon [IFN]-γ in unmanipulated hosts on days 2, 5 or 7 post-infection (p.i.). Three mice per parasite clone were sacrificed at each time-point and plasma was collected for cytokine measurement. These time-points have previously been associated with protective or pathological cytokine responses in P.c.c. AS infections (Li et al. 1999, 2003; Sam and Stevenson, 1999; Su and Stevenson, 2002).
In Exp. 2, the effect of blocking TNFR1 signalling on infection dynamics was investigated via the administration of a soluble TNF Receptor. Soluble TNF Receptor 1 (herein referred to as TNFR-Ig) has been shown to bind and efficiently inhibit mouse TNF-α activity in vivo (Ashkenazi et al. 1991) and was kindly provided by the Therapeutic Antibody Centre, University of Oxford (Oxford, UK). Malaria infections were initiated as in Exp. 1 and mice were intra-muscularly injected with 75 μg of either TNFR-Ig or an isotype control human purified IgG1 (Sigma) dissolved in sterile PBS (Gibco) on days 5, 6, 7 and 8 p.i. (5 mice per clone, per antibody treatment). Data from control mice in Exp. 2 replicated results from Exp. 1. TNFR-Ig treatment was started on day 5 p.i. in order to minimize interference with the anti-parasitic effect of TNF-α early in infection (Langhorne et al. 1989; Stevenson and Tam, 1993).
Monitoring parasitaemia and virulence
Thin blood smears were made daily from tail blood and, after Giemsa staining, the proportions of RBCs parasitized (parasitaemia) were counted microscopically using ×1000 magnification. RBCs were counted until 20 parasites were detected in 2 or more separate fields of vision. This method has been shown mathematically to eliminate the bias in estimates of parasitaemias which arises for statistical reasons when parasites are rare on smears (Crooks, 2004). RBC densities were obtained by flow cytometery (Beckmann Coulter) from a 1[ratio ]40000 dilution of a 2 μl sample of tail blood in Isoton solution every day p.i. in Exp. 1 and on days 0, 2, 4, days 5–12 and then every 2 days until day 21 p.i. in Exp. 2, when mice were sacrificed. Mouse weights were recorded to the nearest 0·1 g on those days p.i. indicated above.
Plasma cytokine detection
In Exp. 1, blood from individual mice was collected into heparinized tubes on the day of sacrifice (days 2, 5 or 7 p.i.). In Exp. 2, 50 μl of tail-blood from individual mice was collected into 50 μl heparin (Sigma) on day 7 p.i. Blood was kept on ice and centrifuged at 500 g for 10 min (4 °C) to obtain plasma, which was frozen at −20 °C until the day of assay. Plasma levels of the cytokines TNF-α, IL-12p70, IL-6, IFN-γ and IL-10 were detected using a flow cytometric cytokine bead array (BD™ CBA Array), with slight modifications from the manufacturer's instructions (BD Biosciences). Briefly, 50 μl of plasma sample or standard were incubated in flat-bottomed 96-well plates (Costar®) with 25 μl of cytokine capture bead mixture (anti-cytokine-coated microspheres) in darkness, with shaking for 1 h at room temperature. Wells were then washed with 200 μl of wash buffer (1×PBS solution) and plates were spun at 200 g for 5 min. Samples and standards were incubated with 25 μl of PE detection reagent (phycoerythrin-conjugated anti-mouse cytokine antibodies) in darkness for 1 h. Wells were then washed and beads re-suspended in 200 μl of wash buffer and analysed on FACsArray analyser (BD™ Biosciences).
Statistical analysis
Traits fell into 3 categories: virulence traits (live-weight and anaemia), parasite traits (parasitaemia) and immunological traits (plasma cytokines). The minimum live-weight and RBC counts reached over the course of infection were used as virulence parameters for analysis of both experiments. Parasite load was quantified as the maximum asexual parasitaemia. In Exp. 1, parasite and virulence data from only the mice that experienced the full course of infection were analysed. Maximum plasma cytokine concentrations were analysed for all mice in that experiment. In Exp. 2, parasitaemia and anaemia observed after the cessation of treatment (days 9 through 16 p.i. inclusive) were also analysed and are referred to as the average post-treatment RBC count or parasitaemia. Finally, immunological data from Exp. 2 (day 7 plasma cytokine concentrations) were analysed. Prior to statistical analysis, it was necessary to transform the data to meet the necessary normality and homogeneity-of-variances assumptions (Grafen and Hails, 2002). Box-Cox transformations were carried out on all non-normal data as an exploratory tool to help determine the optimal transformation required for normalization. In Exp. 1, all cytokine and RBC density data were log10 transformed prior to analysis, whereas in Exp. 2, the weight, IL-12p70 and IFN-γ data were square root transformed, the parasitaemia, IL-6 and IL-10 data were natural log transformed and the RBC parameters were inverse square root transformed. Wherever possible, data are presented in their original units for intuitive ease.
All traits mentioned above were analysed using Analysis of Variance (ANOVAs) or Covariance (ANCOVAs) in MINITAB (release 14, MINITAB Inc.). Explanatory variables for clone, treatment and an interaction between those terms were fitted to the data. Clone had up to 5 factor levels (CW, AS, BC, AJ and Naïve) depending on the analysis, and in Exp. 2, treatment had 2 factor levels (huIgG1 and TNFR-Ig). For all models, we first fitted the maximal model including covariate when relevant, and minimal models were obtained by removing non-significant terms (P-value >0·05). We report F values, the test statistic for ANOVA, as well as P values for full statistical disclosure (Olsen, 2003). Naïve (uninfected) mice suffered significantly less virulence and induced significantly lower cytokine titres relative to infected mice groups regardless of clone, and so were removed from the analyses. To control for the role parasite load may play in determining virulence, a separate analysis was carried out which included maximum parasitaemia as a covariate. Finally, we tested the directional hypothesis that clone virulence increases with increasing pro-inflammatory response (TNF-α or IFN-γ:IL-10) using an ordered heterogeneity test (Rice and Gaines, 1994). The OH test combines the P value from the appropriate ANOVA (Pc) with the Spearman's rank correlation coefficient (rs) to calculate the test statistic (rsPc) as follows: rsPc=r× (1−P) (Rice and Gaines, 1994).
RESULTS
Experiment 1: clone-specific induction of TNF-α.
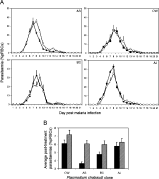
Wild type C57BL/6 female hosts were infected with 106 parasites of 1 of 4 genetically distinct P.c.c. clones; denoted CW, AS, BC and AJ. One mouse died during the experiment and was removed from the analyses (AJ infected mouse on day 13 p.i.). The course of these distinct primary infections, including the virulence and cytokine responses they induced, were examined. Maximum asexual parasitaemias did not differ significantly between clones (Fig. 1A; clone: F3,8=1·7, P>0·05). However, clone differences in virulence were found, with clones AJ and BC inducing lower minimum weight and RBC densities than CW or AS infections (Fig. 1B; clone: F3,7=11, P<0·01; and Fig. 1C; clone: F3,8=6, P<0·05, respectively). These clone differences in weight and RBC loss agree with previous studies (Mackinnon and Read, 1999a,b, 2004) and were maintained even when controlling for parasite load (clone: F3,6=10, P<0·01; and clone: F3,7=10, P<0·01, respectively). Hence, the virulence differences observed between clones are not simply a consequence of parasite burden.

Fig. 1. Effect of Plasmodium chabaudi parasite clone on the kinetics of asexual parasitaemia (A), minimum weight (B) and minimum RBC density (C) reached during genetically distinct single-clone infections. Infections were initiated with 106 RBCs parasitized with 1 of P.c.c. clones CW, AS, BC or AJ. Parasitaemia and virulence measurements were collected every day. Mice were sacrificed on days 2, 5, 7 and 14 p.i. Each line represents the mean of 12, 9, 6 and 3 mice up to these time-points, respectively (+/−S.E.) (A) and each bar represents the least square mean of 3 mice (+/−S.E.) (B and C).
Clones differed in the cytokine profile they induced over time, with significant clone differences in day 5 IFN-γ levels, as well as day 7 TNF-α and IL-6 levels (Fig. 2A; clone: F3,8=14, P<0·001; Fig. 2B; clone: F3,8=5·4, P<0·05 and Fig. 2C; clone: F3,8=7·2, P<0·05). The most virulent clones, AJ and BC, induced high concentrations of pro-inflammatory cytokines: both induced TNF-α; BC also induced IFN-γ, while AJ induced IL-6 (Fig. 2A,B and C). A trend was observed whereby IL-10 levels were elevated during infection with avirulent clones on day 5 p.i., but this was not significant (Fig. 2D; clone: F3,8=1·2, P<0·4). Although clone differences in plasma cytokine profiles were found, only the amount of TNF-α induced on day 7 p.i. correlated with clone differences in virulence (Fig. 2A; clone: F3,8=5, P<0·05, rsPc=0·97, P<0·01; and controlling for average parasitaemia; clone: F3,7=6, P<0·05, rsPc=0·97, P<0·01). Thus, it is possible that clone differences in TNF-α induction around the peak of parasitaemia can help explain clone differences in virulence that are not accounted for by parasite load.

Fig. 2. Effect of Plasmodium chabaudi parasite clone (CW, AS, BC or AJ) on the levels of plasma TNF-α (A), IFN-γ (B), IL-6 (C) and IL-10 (D). Single-clone P.c.c. infections were initiated and blood was obtained from individual mice on the day of sacrifice (days 2, 5 and 7 p.i.). Plasma cytokine levels were determined using a flow cytometric cytokine bead array (see Materials and Methods section). Each bar represents the mean of 3 mice (+/−S.E.).
Experiment 2: effect of blocking TNFR1 signalling during genetically distinct malaria infections
TNFR1 signalling was blocked via the administration of a soluble TNF receptor 1 fusion protein (TNFR-Ig) during each of four distinct P.c.c. infections (as above) and in vivo virulence, parasite dynamics and host plasma cytokine response were followed throughout infection. Three mice died during the experiment and were removed from our analyses (2 IgG-treated, AJ-infected mice and 1 TNFR-Ig-treated, BC-infected mouse on days 10 and 11 p.i.).
(i) Virulence
In vivo blocking of TNFR1 signalling protected mice against malaria-induced weight loss (Fig. 3A,B; treatment: F1,28=7, P<0·01). As observed above, clone differences in weight loss were found (Fig. 3B; clone: F3,28=13, P<0·001), but the level of protection conferred by treatment did not vary among clones (Fig. 3B; clone×treatment: F3,28=2, P=0·2). For example, the protection from weight loss the TNFR-Ig treatment afforded to AS infected mice did not differ significantly from that afforded to AJ infected mice, even though the latter lost almost 2 g more weight. Although clone differences in post-treatment anaemia were found (Fig. 3C and D; clone: F3,28=3, P<0·05), in contrast to weight loss, blocking TNFR1 signalling did not affect the degree of anaemia induced (Fig. 3D; treatment and clone×treatment: P>0·05 in all cases). Thus, blocking TNFR1 signalling protects against malaria-induced weight loss, but the extent of this protection does not vary among clones. In addition, blocking TNFR1 signalling has no effect on the anaemia induced by any of the clones.

Fig. 3. Effect of parasite clone and TNFR-Ig treatment on the kinetics of malaria-induced weight and red blood cell loss. Daily or every other day, mice were weighed and red blood cell counts obtained. Changes in live-weight over time (A), minimum live-weight (B), kinetics of RBC density over time (C), and minimum RBC density (D) during the course of primary infection with 1 of 4 P.c.c. clones (CW, AS, BC or AJ) are shown for TNFR-Ig treated mice (open symbols and hatched bars) or control IgG-treated mice (solid symbols and solid bars). Each line or bar represents the mean of 5 mice (+/−S.E.), except where deaths occurred as noted in the Results section.
(ii) Parasite load
For the first 8 days of infection, treatment did not significantly affect asexual parasite dynamics (Fig. 4A). However, after treatment stopped on day 9 p.i., depleted mice suffered elevated parasitaemias (Fig. 4A and B; average post-treatment parasitaemia; treatment: F1,29=7, P<0·05). We analysed several aspects of the parasite data presented in Fig. 4A. While there were clone differences in average parasitaemia and day of peak parasitaemia (Fig. 4A; clone: F3,29=5, P<0·01; and clone: F3,29=10, P<0·001, respectively) and near significant differences in both the maximum and average post-treatment parasitaemia (clone: F3,29=2, P=0·09; and clone: F3,29=3, P=0·06, respectively), the magnitude of these clone differences was unaffected by treatment (clone×treatment: P>0·05 in all cases). Thus, blocking TNFR1 signalling had similarly negligible effects on parasite kinetics for all the clones.

Fig. 4. Effect of parasite clone and TNFR-Ig treatment on the kinetics of asexual parasitaemia. Plots represent the percentage parasitized RBCs during infection with 1 of 4 P.c.c. clones (CW, AS, BC or AJ) in TNFR-Ig-treated mice (open symbols and hatched bars) or control IgG-treated mice (solid symbols and filled bars) (A). Bars represent the least square mean of post-treatment asexual parasitaemia (days 9–16 inclusive), broken down by clone and treatment (B). Each line or bar represents the mean of 5 mice (+/−S.E.), except where deaths occurred as noted in the Results section.
(iii) Per-parasite virulence
To differentiate between the roles that parasite load and immunopathology play in driving malaria virulence (Graham et al. 2005) we examined the effects of clone and treatment on virulence by statistically taking maximum parasitaemia into account. Although parasite load was a significant predictor of weight loss (F1,27=8, P<0·01), it did not affect clone differences in weight loss, nor the protective effect of treatment on minimum weight (clone: F3,27=10, P<0·001, treatment: F1,27=7, P<0·05, clone×treatment: F3,27=2, P=0·1). These results show that TNFR-1 signalling contributes towards malaria-induced weight loss, independently of parasite load or parasite genotype. Thus, the virulence differences among clones and treatments depicted in Fig. 3B and D are not due to parasitaemia differences alone.
For a given parasite burden, TNFR-Ig treated mice suffered greater anaemia from day 9 p.i. onwards after treatment ended (treatment: F1,27=5, P<0·05). In contrast, clone differences in post-treatment anaemia were removed once parasite load was taken into account (clone: F3,27=2, P=0·2 and clone×treatment: F3,27=0·4, P=0·8). Parasite load alone thus explains clone differences in post-treatment anaemia.
(iv) Cytokines associated with immunopathology
To examine the induction of cytokines that might contribute to the clone or treatment effects on virulence, we measured the levels of several plasma cytokines on day 7 p.i. during Exp. 2. Clones differed significantly in the induction of plasma IFN-γ, IL-6 and IL-10 on day 7 p.i. (Table 1). Treatment, on the other hand, only significantly affected plasma IL-6, with TNFR-1 blockade causing elevated IL-6 levels on day 7 p.i., regardless of P.c.c. clone (Table 1).

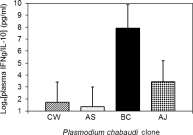
We are interested in whether clone virulence or avirulence relates to the development of pro- or anti-inflammatory cytokine responses. In this regard, we found that controlling for parasite load, day 7 plasma IFN-γ levels helped to explain both the minimum live-weight and anaemia reached (F1,26=21, P<0·001 and F1,26=8, P<0·01, respectively). Just as we investigated the role that parasite load plays in driving virulence in the previous section, we now observe that our virulence results (Fig. 3B,D) relate to IFN-γ levels. Thus, elevated IFN-γ around the peak of infection may have detrimental virulence outcomes (Kremsner et al. 1992; Waki et al. 1992). High ratios of pro- to anti-inflammatory cytokines have been associated with malaria virulence in the field (Dodoo et al. 2002). In agreement with this, our data show that the ratio of IFN-γ to IL-10 is higher for more virulent P.c.c. infections, regardless of parasite load (Fig. 5; clone: F3,28=3·2, P<0·05; and clone: rsPc=0·966, P<0·01). Thus, it is possible that clone differences in IFN-γ induction, or in the regulation of IFN-γ by anti-inflammatory cytokines, contribute towards clone differences in malaria virulence.

Fig. 5. Effect of parasite clone on the ratio of plasma IFN-γ to IL-10 on day 7 post-infection, for each of 4 P.c.c. clones (CW, AS, BC or AJ). Blood was obtained from individual mice on day 7 p.i. and plasma levels of IFN-γ and IL-10 were determined. The natural log transformation was applied to data to meet the necessary homogeneity-of-variance and normality assumptions. Bars are least square mean of 5 mice per infecting clone (±S.E.), except where deaths occurred as noted in the Results section.
DISCUSSION
Malaria virulence may be induced directly by the parasite or via immune-mediated mechanisms, which include TNF-α ( Grau et al. 1987; Clark and Chaudhri, 1988a,b; Kern et al. 1992). For example, high levels of serum TNF-α correlate with poor outcome and disease severity in both rodent (Clark and Chaudhri, 1988a,b) and human (Kwiatkowski, 1990; Kwiatkowski et al. 1990) malaria infections. We hypothesized that P.c.c. clones may achieve high virulence by (i) growing to high parasite densities, (ii) inducing an exacerbated TNF-α response, or (iii) via a combination of these factors.
In Exp. 1, we observed that more virulent malaria clones induced higher day 7 plasma TNF-α levels, even when parasite load was taken into account. In Exp. 2, TNFR1 signalling was blocked during genetically distinct infections, with the expectation that clone virulence schedules would be differentially affected and possibly eliminated. However, we found that regardless of parasite genotype, blocking TNFR-1 signalling ameliorated weight loss, and had little effect on anaemia or parasitaemia. These data suggest that the contribution of TNF-α to malaria virulence does not depend on P.c.c. genotype. Thus, the previously reported effects of TNF-α on both the virulence and parasitaemia during P.c.c. AS infections (Jacobs et al. 1996; Li and Langhorne, 2000; Sam et al. 1999) appear representative of those observed across the suite of P.c.c. clones we studied.
Separating the pathogenic effects of an overzealous immune response from those of parasite burden is extremely difficult (Graham et al. 2005). By adding parasitaemia to statistical models, we were able to show that clone differences in the induction of plasma TNF-α on day 7 p.i. are not completely driven by parasite load and correlate with virulence. In the same respect, lethal and non-lethal P. yoelii infections have been shown to differ in their ability to induce early TGF-β, despite no significant differences in parasite load at that time (Omer et al. 2003). Furthermore, the enhanced disease severity of IL-10-deficient mice infected with the parasites Trypanosoma cruzi (Hunter et al. 1997), Toxoplasma gondii (Gazzinelli et al. 1996), Helicobacter hepaticus (Kullberg et al. 1998) or P. chabaudi (Li et al. 1999) did not correlate with increased parasite burdens, but rather with an increased inflammatory response. These data suggest that for many parasite species, disease severity is at least partly immune mediated and independent of parasite load.
There are several possible mechanisms by which different malaria genotypes might induce TNF-α, independent of parasite burden. For example, it is plausible that the cellular source of TNF-α, or alternatively the trigger for TNF-α release, may differ during infections with genetically distinct parasites. It has been shown that during P.c.c AS infections, macrophages (Stevenson et al. 1992) and dendritic cells (Seixas et al. 2001) can produce TNF-α upon direct stimulation by the parasite in vitro, and CD4+ T cells have also been shown to produce TNF-α during malaria infection (Hirunpetcharat et al. 1999). If P.c.c. clones differ in ability to induce these cellular responses, this could explain the differential amplification of TNF-α we observe in the plasma. It would be of interest to determine what the cellular source(s) of cytokines are during infection with our distinct clones, and also to examine whether clones differ in antigenicity. A substantial amount is currently known regarding P.c.c. AS immunology (Langhorne et al. 2004; Stevenson and Riley, 2004), but all other P.c.c. clones await similar attention.
Our data suggest that TNFR1 signalling is dispensable (between days 5 and 8 p.i.) for the ultimate control of malaria infection, regardless of parasite genotype. To our knowledge, the reagents used in this study do not distinguish between TNF-α and the closely related cytokine, lymphotoxin-α (LT-α), which can both signal through TNFR1 and thus we have been cautious to attribute treatment effects to TNFR1 signalling rather than TNF-α itself. In any case, cytokine redundancies may help explain the relatively small effects of TNFR-Ig treatment. For example, IL-6 or IFN-γ could be compensating for the absence of TNFR1 signalling. It has been proposed that differences in induction of key anti-inflammatory cytokines, which act to down-regulate the nascent pro-inflammatory response, may also contribute to malaria pathology (Dodoo et al. 2002; Artavanis-Tsakonas et al. 2003). In this regard we found that ratios of plasma IFN-γ to IL-10 correlate with clone virulence. Future experiments designed to block other key cytokines (including IL-6 and IFN-γ), possibly in combination, during diverse malaria infections may reveal cytokines that differentially affect clone virulence. Finally, it is possible that TNFR-independent pathways for virulence determination exist, for example both the induction of nitric oxide (NO), and generation of reactive oxygen species have been implicated in the pathogenesis of severe malaria (Clark et al. 1983; Anstey et al. 1996; Griffiths et al. 2001), and may contribute to the clone virulence differences observed.
In conclusion, our results demonstrate that blocking TNFR1 signalling protects against weight loss during P.c.c. infections, regardless of parasite genotype or parasitaemia. However, a role for parasite genotype in determining the contribution of IFN-γ and IL-6-mediated pathology to virulence is not ruled out by this study. Hence, studies aimed at investigating the contribution of cytokine-mediated pathways to the virulence induced by genetically diverse malaria infections deserve further attention. Clearer definition of the root causes of malarial virulence, including the proportion due to immunopathology, would help to identify essential targets which could be used in the treatment of malaria.
G.H.L. is supported by a Wellcome Trust studentship and A.L.G. is supported by the Leverhulme Trust and the School of Biological Sciences, University of Edinburgh. We thank the staff of the March House, University of Edinburgh, for excellent animal husbandry, Dr P. Bird (Therapeutic Antibody Centre, University of Oxford, UK) for providing TNFR-Ig, S. MacCall for technical assistance in performing CBA assays and H. Ferguson for helpful comments on the manuscript. This work was supported by the Wellcome Trust (069299/Z/02/A).