



Introduction
The global prevalence of type 2 diabetes (T2D) has been increasing rapidly and is now one of the major public health challenges worldwide. While it has been thought that the development of T2D is a result of aging, genetics, and lifestyle factors, it is increasingly accepted that T2D susceptibility is established in utero. Reference Bruce1 Moreover, early life exposures to chemical insults may also play a key role in the development of T2D in postnatal life. Reference De Long and Holloway2 There is evidence to suggest that exposure to nicotine during pregnancy increases the risk of metabolic disorders, including T2D, in the offspring. Reference Bruin, Gerstein and Holloway3
T2D is characterized by peripheral insulin resistance and beta-cell dysfunction. Furthermore, in rodents, early life exposure to nicotine leads to decreased beta-cell mass, increased beta-cell apoptosis, and impaired glucose homeostasis similar to what is observed in T2D. Reference Bruin, Gerstein and Holloway3 Interestingly, several studies have implicated p66shc, a member of the ShcA (Src homologous-collagen homolog) adaptor protein family, in mitochondrial dysfunction, oxidative stress, and apoptosis. Reference Migliaccio, Giorgio and Mele4 Specifically, activation of p66shc has been shown to lead to reactive oxygen species (ROS) production and mitochondrial-induced apoptosis, Reference Migliaccio, Giorgio and Mele4 comparable to what has been reported in the pancreas following perinatal exposure to nicotine. Reference Bruin, Petre, Raha, Morrison, Gerstein and Holloway5
There are several lines of evidence to suggest that the pancreatic deficits reported in nicotine-exposed offspring may be mediated via p66shc. First, Oke et al Reference Oke, Sohi and Hardy6 have reported increased p66shc expression in animals with growth restriction followed by postnatal catch-up growth; nicotine-exposed animals exhibit a similar growth trajectory. Reference Holloway, Lim, Petrik, Foster, Morrison and Gerstein7 Second, there is evidence that p66shc can directly influence beta-cell apoptosis; overexpression of p66shc in INS-1E cells enhanced glucose and palmitate-stimulated apoptosis, an effect that was partially blocked by transfection with p66shc siRNA. Reference Natalicchio, Tortosa and Labarbuta8 In addition, activation of p66Shc in INS-1E cells increased mitochondrial ROS production, a key player in beta-cell apoptosis. Reference Karunakaran, Lee, Elumalai, Moon and Won9 Finally, studies have shown that exposure to nicotine alone in vivo Reference Arany, Clark, Reed and Juncos10 or as a component of cigarette smoke in vitro Reference Zhang, Tang and Shan11 can increase the expression of p66shc in the kidney and in airway epithelial cells, respectively. The goal of this study was to examine the effects of nicotine in vivo and in vitro on pancreatic p66shc expression. Furthermore, as p66shc expression has been demonstrated to be influenced by posttranslational histone modifications Reference Zhou, Chen and Wan12 and nicotine has been shown to increase the transcription of p66shc via epigenetic mechanisms, Reference Arany, Clark, Reed and Juncos10 we also examined the effect of nicotine on epigenetic histone modifications including markers of both histone demethylation (lysine demethylase 4A (Kdm4a) and 4C (Kdm4c)) and histone methylation (suppressor of variegation 3–9 homolog 1 (Suv39h1) and 2 (Suv39h2)) and ASH1-like histone lysine methyltransferase (Ash1l)) in the neonatal pancreas.
Methods
Animal maintenance and treatment
All procedures were performed in accordance with the guidelines of the Canadian Council of Animal Care. Nulliparous 200–250 g female Wistar rats (Envigo, Indianapolis, IN) were maintained under controlled lighting (12:12 L:D) and temperature (22°C) with ad libitum access to food and water. Two weeks prior to mating, the dams were randomly assigned to receive either saline (vehicle) or nicotine. Dams (N = 10 per group) were injected with saline or 1.0 mg/kg/day nicotine bitartrate (Sigma-Aldrich, St. Louis, MO) subcutaneously for 14 d prior to mating, and during pregnancy. Dams were allowed to deliver normally, and pups were euthanized at postnatal day 1 (PND1) for pancreas tissue collection. The dose of nicotine used in this animal model results in maternal serum cotinine concentrations of 136 ng/ml, Reference Holloway, Kellenberger and Petrik13 which is within the range of cotinine levels reported in women who are considered “moderate smokers” (80–163 ng/ml), Reference Eskenazi and Bergmann14 and serum cotinine concentrations of 26 ng/ml in the nicotine-exposed offspring at birth, Reference Holloway, Kellenberger and Petrik13 which is also within the range (5–30 ng/ml) observed in infants nursed by smoking mothers. Reference Luck and Nau15 Pancreas tissue was collected from two randomly selected pups per liter, one for RNA isolation and the other for histone extraction and quantification. Tissues were excised, flash frozen, and stored at −80°C. Total RNA was then isolated and extracted using TRIzol reagent (Invitrogen, Carlsbad, CA).
Cell culture maintenance and treatment
INS-1E rat insulinoma pancreatic beta cells (generously provided by Dr. Claes Wollheim, University of Geneva, Geneva, Switzerland) were cultured in RPMI-1640 media containing 10 mM 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) and 2 mM L-glutamine (RPMI; Corning, Manassas, VA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco, Grand Island, NY), 50 μM 2-mercaptoethanol (EMD Millipore, Etobicoke, ON), 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) at 37°C in a humidified atmosphere of 95% O2 and 5% CO2.
Cells were treated for 48 h with 0 (vehicle control) and 1 μM nicotine bitartrate (Sigma-Aldrich) diluted in supplemented RPMI as described above. This concentration was selected based on prior work showing impaired insulin secretion and mitochondrial dysfunction in INS-1E cells at this dose. Reference Woynillowicz, Raha, Nicholson and Holloway16,Reference Yoshikawa, Hellström-Lindahl and Grill17 After 48 h, cells were washed with phosphate-buffered saline and total RNA was then isolated and extracted (N = 7–8 independent experiments) using TRIzol reagent (Invitrogen). Additional cells were collected and frozen at −80°C for histone extraction and quantification (N = 5–7 independent experiments).
RNA isolation and real-time quantitative polymerase chain reaction (qPCR)
Total RNA concentration from PND1 pancreas tissue and INS-1E cells was determined using the Nanodrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA). Complementary DNA (cDNA) was synthesized from 2 μg of RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) as per the manufacturer’s instructions. The resulting cDNA was used as a template for qPCR, which was performed using PerfeCTa® SYBR® Green FastMix® (Quanta Biosciences, Gaithersburg, MD) on the LightCycler® 480 (Roche, Mississauga, ON). To explore expression of p66shc and methylation drivers in histone H3 lysine 9 (H3K9) methylation, we quantified p66shc and markers of both histone demethylation and histone methylation. For genes involved in H3K9 demethylation, we selected lysine demethylase 4A (Kdm4a) and 4C (Kdm4c). Reference Marmorstein and Trievel18,Reference Su, Wang and Lee19 For genes involved in H3K9 methylation, we selected suppressor of variegation 3–9 homolog 1 (Suv39h1) Reference Costantino, Paneni and Virdis20 and 2 (Suv39h2) Reference Lehnertz, Ueda and Derijck21 and ASH1-like histone lysine methyltransferase (Ash1l). Reference Gregory, Vakoc and Rozovskaia22 The primers were (listed as gene, forward and reverse sequence): p66shc, 5′-GTCCGACTACCCTGTGTTCCTT-3′ and 5′-CAGCAGGATTGGCCAGCTT-3′; Kdm4a, 5′-CTTTAGGCTCCAGGTTGTTCCT-3′ and 5′-TCATCATAGGAGGTTCGAGGC-3′; Kdm4c, 5′-GGTGGAGAGTCCCCTAAATCC-3′ and 5′-GAACTCCCGAAA-CTCCTCCAT-3′; Suv39h1, 5′-TTTCTGTGTACCCTCCTCAGC-3′ and 5′-GAGCAACCCCAGTTAACCTCT-3′; Suv39h2, 5′-AACCTTGATACTCGGCTTCCC-3′ and 5′-TGGCAGGGCTGTAGTCAATAG-3′; and Ash1l, 5′-ACAGACCCAAAGAGCCGATT-3′ and 5′-CAACGTAGACATCAACATGAATGG-3′. The cycling conditions included polymerase activation (95°C for 10 min), followed by 40 cycles of denaturing (95°C for 15 s) and annealing/elongation (60°C for 1 min). Levels of gene expression were calculated using the ΔΔCt method Reference Livak and Schmittgen23 and normalized using the geometric means of two reference genes: hypoxanthine phosphoribosyltransferase 1 (Hprt1) (5′-GCAGTACAGCCCCAAAATGG-3′ and 5′-GGTCCTTTTCACCAGCAAGCT-3′) and peptidylprolyl isomerase A (Ppia) (5′-CCGCTGTCTCTTTTCGCC-3′ and 5′-GCTGTCTTTGGAACTTTGTCTGC-3′).
Acid extraction of histones
Pancreas tissue and INS-1E cell pellets were homogenized with lysis buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, and protease inhibitor tablets; 1:5 ratio tissue pellet:lysis buffer) and HCl added to a final concentration of 0.2 M. Samples were placed on a rocker at 4°C for 1 h and then centrifuged at 11,000g for 10 min in an Eppendorf 5417R centrifuge at 4°C. The acid-insoluble pellet was discarded and the supernatant (approximately 500 µl) was dialyzed in 200 ml of dialysis buffer using the Pierce Slide-A-Lyzer® MINI dialysis units (3500 MW cut-off) in the following manner at 4°C on a rocker: twice in 0.1 M acetic acid for 1 h, once in water for 1 h, once in water for 3 h, and in water overnight. The samples were then aliquoted into separate tubes and stored at −20°C.
Histone quantification and immunoblotting
Protein loading samples were prepared utilizing NuPAGE lithium dodecyl sulfate Sample Buffer NuPAGE Sample Reducing Agent (Invitrogen). Loading amounts were 10–15 µg for histone extracts. Samples were run on NuPage 4%–11.5% Bis-Tris gels (Invitrogen) in 2-(N-morpholino)ethanesulfonic acid (MES) - sodium dodecyl sulphate (SDS) running buffer at 150 V for 1–2 h. After transfer to nitrocellulose membranes, membranes were blocked for 2 h in 5% non-fat dried milk in 0.1% tris buffered saline with Tween 20 (TBST). Membranes were incubated with antibodies targeted against total nuclear histone H3 (EMD Millipore, 05-499), and trimethylated histone H3 (lysine 9, EMD Millipore, 07-422). After washing in 0.1% TBST, membranes were incubated for 2 h with the appropriate secondary antibody (1:10,000 dilution). Bands were normalized to appropriate reference proteins. Beta-actin was probed with antibody (Sigma-Aldrich) for a 2-h room temperature incubation. All bands were detected with a SuperSignal® West Dura Extended Duration Substrate (Thermo Scientific) and imaged with a VersaDoc Imaging System (Bio-Rad). Densitometric analysis on blots was performed with Quantity One Software (Bio-Rad). Data were normalized to reference proteins (beta-actin for cytoplasmic proteins, or total histone H3 for nuclear proteins) by applying stripping buffer (200 mM glycine, 0.1% sodium dodecyl sulfate, and 1% Tween 20) for 10 min, and then the detection protocol was implemented as above.
Statistical analysis
All statistical analyses were performed using SigmaPlot (v.11.2, Systat Software, San Jose, CA). Data were tested for outliers (Grubbs’ test), normality, and equal variance. Means were compared between control and nicotine groups using Student’s t-test. Data that failed normality and/or equal variance were analyzed by using the Mann–Whitney rank sum test. Values are presented as mean ± standard error of the mean (SEM).
Results
In PND1 nicotine-exposed rat offspring, steady-state mRNA levels of pancreatic p66shc were increased compared to control offspring (Fig. 1a). Similarly, nicotine exposure significantly increased p66shc mRNA in INS-1E cells (Fig. 1b).
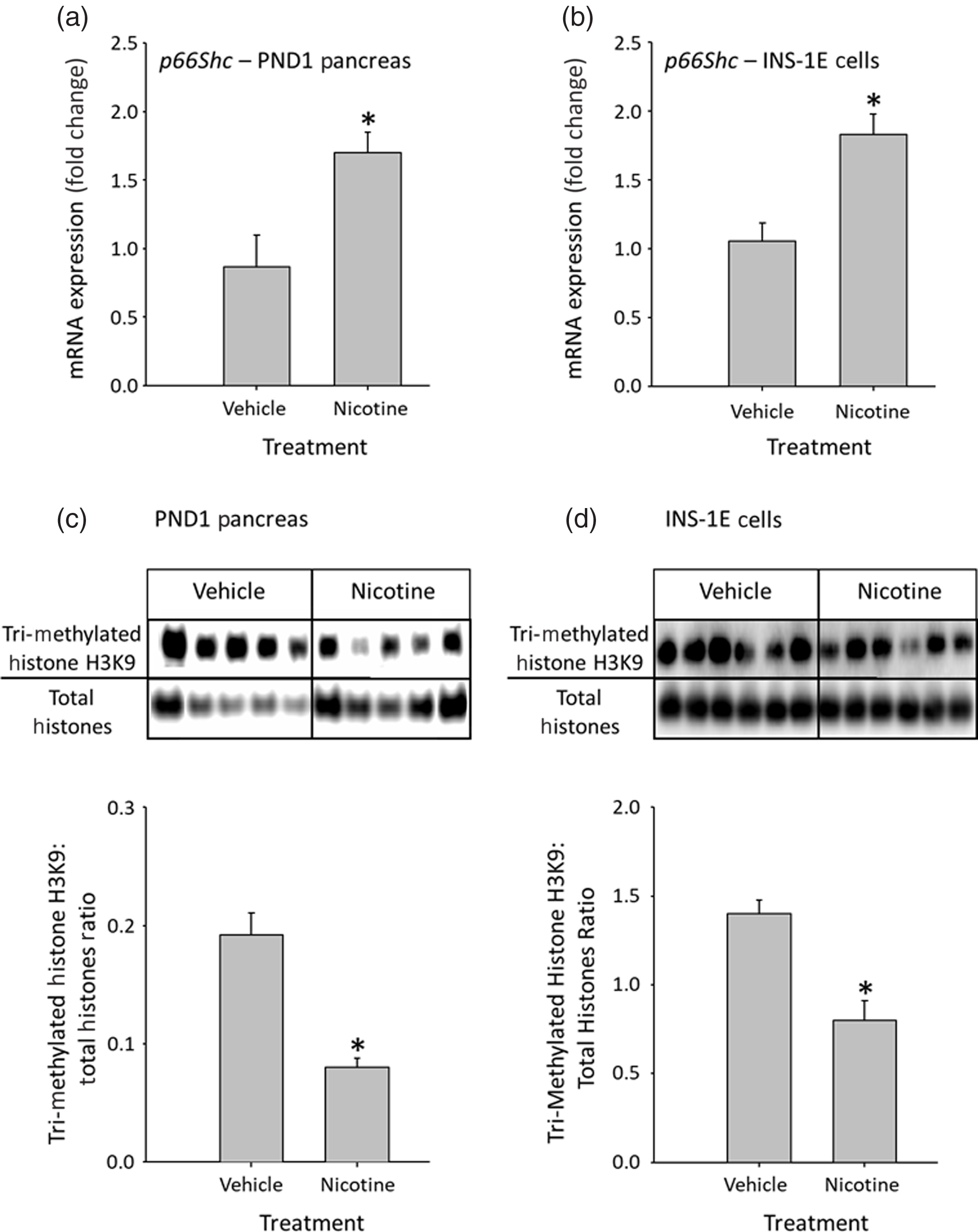
Fig. 1. Effect of nicotine exposure on p66shc mRNA in ( a ) PND1 pancreas and ( b ) INS-1E cells. Effect of nicotine exposure on H3K9 methylation (involved in chromatin silencing) in ( c ) PND1 pancreas and in ( d ) INS-1E cells. Representative histone blots are presented above bar graphs showing the quantification of blot intensity. Data are represented as mean ± SEM, N = 4–9 animals per group (PND1 pancreas) or 5–8 independent experiments (INS-1E cells). Values with asterisks (*) are significantly different to control, P < 0.05.
Given that exposure to nicotine in utero has been demonstrated to lead to posttranslational histone H3 modifications in other organs including decreased H3K9 methylation in the brain, Reference Suter, Abramovici and Griffin24 and that nicotine has been shown to increase the transcription of p66shc via epigenetic mechanisms in vitro, Reference Arany, Clark, Reed and Juncos10 we next investigated if alterations in pancreatic p66shc expression were associated with the methylation of histone H3K9, which is involved in chromatin silencing. Nicotine treatment significantly decreased histone H3K9 methylation in PND1 pancreas (Fig. 1c) and INS-1E cells (Fig. 1d).
To elucidate which specific lysine methylases or lysine demethylases might mediate the decrease in histone H3K9 methylation in the PND1 pancreas, we next measured the expression of key histone methylation modifying enzymes. While the steady-state mRNA levels of lysine demethylase Kdm4a were unchanged in the PND1 nicotine-exposed pancreas (Fig. 2a), the mRNA expression of the lysine demethylase Kdm4c was significantly increased versus control animals (Fig. 2b). There was no effect of nicotine exposure on the mRNA expression of the lysine methylases Suv39h2 and Ash1l (Fig. 2d and 2e), but there was a significant decrease in Suv39h1 expression in nicotine-exposed animals (Fig. 2c).
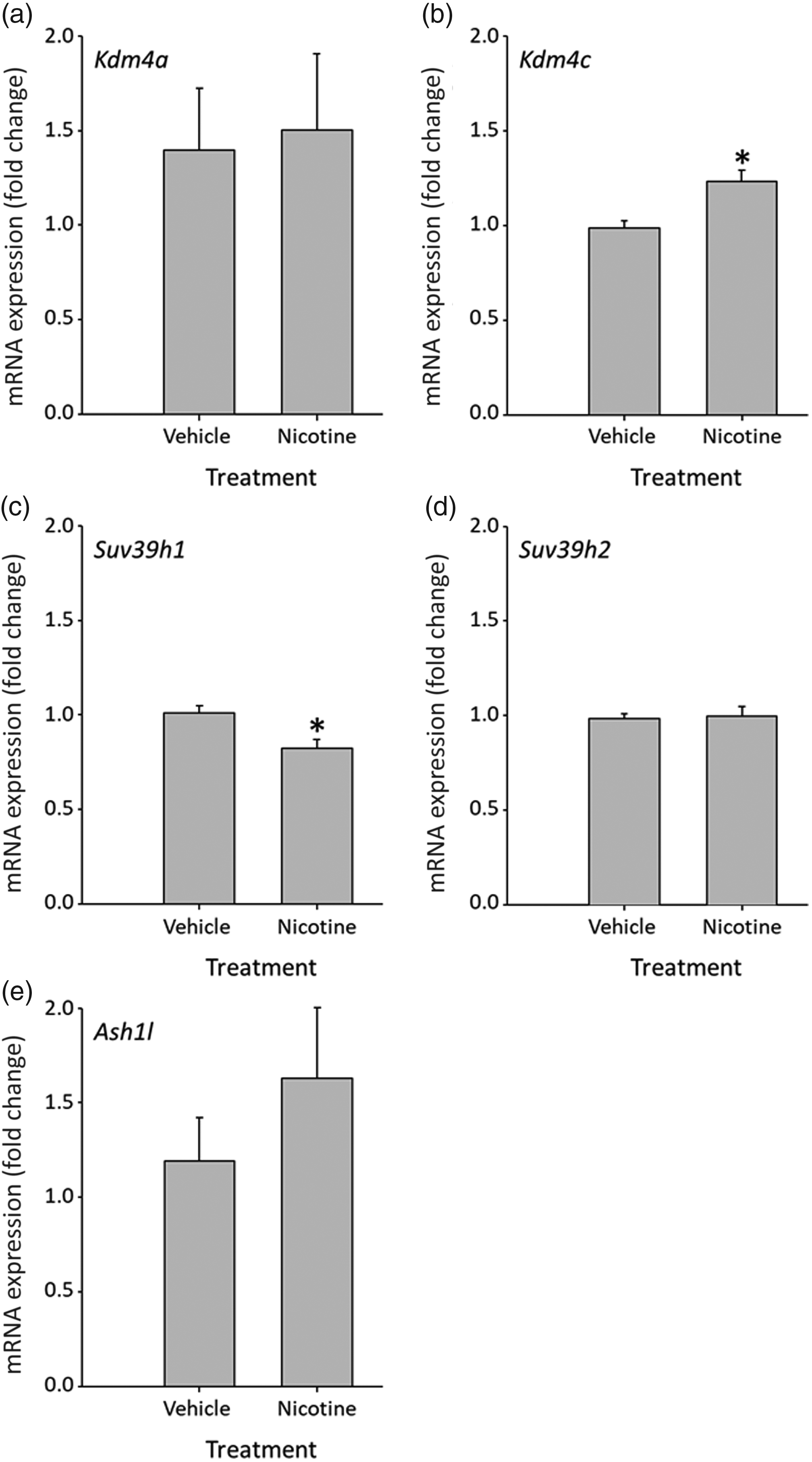
Fig. 2. Effect of prenatal nicotine exposure on expression of ( a ) Kdm4a mRNA (a histone lysine demethylase), ( b ) Kdm4c mRNA (a histone lysine demethylase), ( c ) Suv39h1 mRNA (a histone lysine methyltransferase), ( d ) Suv39h2 mRNA (a histone lysine methyltransferase), and ( e ) Ash1l mRNA (a histone lysine methyltransferase) in PND1 pancreas. Data represented as mean ± SEM, N = 7–10. Values with asterisks (*) are significantly different to control, P < 0.05.
Discussion
This study investigated the effects of maternal nicotine exposure on the expression of pro-oxidative adaptor protein p66shc in the pancreas of nicotine-exposed rat offspring. To reinforce that the effects in vivo are attributable to direct effects of nicotine in the pancreatic beta cells, we confirmed our results in vitro using the INS-1E beta-cell line. Prenatal exposure to nicotine increased mRNA expression of p66shc in the neonatal rat pancreas. Similarly, p66shc expression was increased following nicotine treatment in vitro. The increase in p66shc expression in vivo occurred in association with diminished histone H3K9 methylation. Furthermore, this reduced methylation occurred with changes in the expression of histone-modifying enzymes; specifically, an increase in lysine demethylase and a decrease in lysine methyltransferase. As other studies have demonstrated that elevated p66shc mediates oxidative stress, mitochondrial dysfunction, and cell injury in general including beta-cell apoptosis, Reference Migliaccio, Giorgio and Mele4,Reference Natalicchio, Tortosa and Labarbuta8,Reference Giorgio, Migliaccio and Orsini25 the increased expression of p66shc likely underlies pancreatic deficits reported following early life exposure to nicotine. Reference Bruin, Petre, Raha, Morrison, Gerstein and Holloway5,Reference Holloway, Lim, Petrik, Foster, Morrison and Gerstein7
Given that fetal growth-restricted offspring exhibit higher postnatal hepatic p66shc protein abundance in postnatal life, Reference Oke, Sohi and Hardy6 it is plausible that the increase in p66shc expression in the pancreas of nicotine-exposed animals is indirectly related to nicotine-induced reductions in fetal growth. Reference Holloway, Lim, Petrik, Foster, Morrison and Gerstein7,Reference Ma, Nicholson, Wong, Holloway and Hardy26 However, our in vitro studies using INS-1E cells demonstrate that nicotine can directly increase p66shc expression in pancreatic beta cells. Moreover, a previous study has demonstrated that p66shc expression is upregulated following nicotine exposure in renal proximal tubule cells. Reference Arany, Clark, Reed and Juncos10 As nicotine readily crosses the placenta and is present in breast milk, Reference Luck and Nau15 it is likely that the increased expression of p66shc in the neonatal pancreas was a direct result of exposure to nicotine in utero and during the first day of neonatal life. Upregulation of p66shc leads to pancreatic beta-cell dysfunction, apoptosis, and cytotoxicity, Reference Natalicchio, Tortosa and Labarbuta8 and higher levels of p66shc have been reported in T2D patients, Reference Pagnin, Fadini, de Toni, Tiengo, Calò and Avogaro27 suggesting that this nicotine-induced increase in pancreatic p66shc expression early in postnatal life may be central to the metabolic deficits seen in nicotine-exposed animals in adulthood. Reference Holloway, Lim, Petrik, Foster, Morrison and Gerstein7
The mechanisms by which early life exposure to nicotine can transcriptionally regulate pancreatic p66shc expression are not fully understood, but may involve epigenetic modifications. In renal proximal tubule cells, nicotine increased p66shc transcription via epigenetic modifications. Reference Arany, Clark, Reed and Juncos10 In the present study, it was found that there was a decrease in the ratio of trimethylated histone H3K9 to total histones following nicotine exposure in both PND1 pancreas and INS-1E cells, demonstrating that exposure to nicotine in utero may lead to an active chromatin environment permissive for p66shc transcription. Similarly, prenatal nicotine exposure and adult cigarette smoke exposure in rodents have been shown to result in decreased H3K9 methylation in the brain. Reference Suter, Abramovici and Griffin24 Decreased lysine 9 trimethylation has been shown to promote a euchromatin state, Reference Marmorstein and Trievel18 which is consistent with our observed increased p66shc mRNA expression.
It has been suggested that the histone modifications that occur in utero are sustained by a diverse range of histone-modifying enzymes whose expression levels are governed by external environmental insults (i.e., drugs, hypoxia, and low nutrients) during key perinatal windows of development. Reference Marmorstein and Trievel18 With respect to histone H3 methylation, histone-modifying enzymes exist to either methylate or demethylate the lysine 9 residue, ultimately influencing overall gene transcription. Reference Marmorstein and Trievel18 In the nicotine-exposed PND1 pancreas, we observed an increase in the steady-state mRNA levels of the histone lysine demethylase Kdm4c and a decrease in the expression of the histone lysine methyltransferase Suv39h1. Collectively, these changes have the potential to promote a decrease in histone H3K9 methylation resulting in a euchromatin environment. It is noteworthy that, in mutant obese C57BL/6 mice, Suv39h1 expression decreased concomitantly with an increase in Kdm4c expression, resulting in lower histone H3K9 methylation of the p66shc promoter and a corresponding increase in p66shc gene expression. Reference Costantino, Paneni and Virdis20
Taken together, results of this study suggest that nicotine exposure during fetal development can increase the expression of p66shc in the neonatal pancreas via histone modifications. As p66shc is associated with mitochondrial oxidative stress and beta-cell apoptosis, Reference Natalicchio, Tortosa and Labarbuta8 an increase in p66shc provides a potential link between in utero nicotine exposure and beta-cell dysfunction in postnatal life, and may be a potential therapeutic target to ameliorate the long-term metabolic deficits seen in children born to women who smoke during pregnancy.
Acknowledgements
The authors would like to thank Eric Barry, Peter Gariscsak, Catherine Nicholson, and Dr. Michael Wong for technical assistance with this project.
Financial support
This work was supported by a grant to ACH from the Canadian Institutes of Health Research (PJT-155981) and a Natural Sciences and Engineering Research Council Discovery Grant (RGPIN 04164) to DBH.
Conflicts of interest
None.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guides on the care and use of laboratory animals (Canadian Council of Animal Care) and have been approved by the institutional animal care committee at McMaster University.


