


Nerve agents are chemicals that inhibit the enzyme acetylcholinesterase, resulting in symptoms of a muscarinic cholinergic crisis and convulsionsReference Eddleston, Buckley, Eyer and Dawson1 (Table 1). Scenarios involving nerve agent exposure can range from a small agricultural occupational exposure to a large terrorist attack, such as the 1995 release of sarin in the Tokyo subway system.Reference Zwiener and Ginsburg2, Reference Tokuda, Kikuchi, Takahashi and Stein3 The diversity of scale presents challenges in emergency planning at the community level.
Table 1 Symptoms of Nerve Agent Toxicity*

*This list of symptoms, the so-called DUMBBBELS, is often preferred to SLUDGE because it highlights the life-threatening symptoms of bradycardia, bradypnea, and bronchorrhea.
One challenge facing emergency planners and providers is the need to have each antidote immediately available. Contingency strategies are also needed when the supply of first-line antidotes run out. For example, current medical countermeasures (MCMs) for the treatment of patients exposed to a nerve agent include oxime agents, such as pralidoxime; antimuscarinic agents, such as atropine; and anticonvulsants, primarily agents from the class of gamma aminobutyric acid (GABA) receptor-agonist benzodiazepines.Reference Eddleston, Buckley, Eyer and Dawson1 Stockpiles of Food and Drug Administration (FDA), currently approved MCMs – pralidoxime and atropine – in the prehospital setting are supplied as a single-dose mono- or dual-chambered auto-injector (AI).4, Reference Hall and Walter5 AIs in municipal caches are of limited supply due to cost, stockpile maintenance, competing antidotal priorities, and sourcing challenges. In a large nerve agent exposure, once the AI stockpile has been exhausted, the administration of first-line MCMs (ie, atropine, pralidoxime, and diazepam) must rely on remaining multidose vial stocks and temporizing parenteral routes such as intramuscular, intraosseous, and intravenous.
This review is not a governmental endorsement of off-label uses of medications; rather, it is a review of the efficacy and pharmacokinetic literature supporting the Department of Homeland Security (DHS) working group’s subject matter expert consensus. We aim to inform community emergency planners, first responders/first receivers, and local medical authorities of alternative pharmaceutical options when first-line treatments are no longer available. This article does not address non-pharmaceutical MCMs, prophylactic treatment strategies, neuroprotective agents, personal protective equipment, or patient decontamination. All are complementary protective actions to timely pharmaceutical antidote administration and are essential elements of an optimized and ultimately successful response to a community with a mass nerve agent exposure incident.
METHODS
The DHS Chemical Defense Program convened an ad hoc expert working group. The working group consisted of experts from the fields of medical toxicology, pharmacy, emergency medicine, chemistry, emergency medical services, neurology, and pediatrics. The initial 2-day working group meeting was facilitated by cognitive-science contractors versed in the development and fielding of decision-support tools and resource and operational solutions for emergency response. A subsequent review of the symposium output, combined with a comprehensive literature review focusing on the specific literature that met the federal government’s Public Health Emergency Medical Countermeasures Enterprise (PHEMCE) Product Specific Requirements for nerve agent MCM research and development, was performed. This work was crafted into several federal interagency presentations and a working group methodology and summary document. To address literature gaps in comparative efficacy against nerve agents and pharmacokinetic data for contingency routes of administration (such as intranasal), specific research projects were nominated to the United States Army Research Institute for Chemical Defense (USAMRICD, Edgewood, MD) and the Biomedical Advanced Research and Development Agency (BARDA/Health and Human Services) for further study. The draft working group summaries were reviewed by the American College of Medical Toxicology as well as the federal Chemical Intergovernmental Planning Team and the developers of the nationally recognized Advanced Hazmat (hazardous materials) Life Support (AHLS) course for first responders.
Alternative MCMs were included only if the proposed pharmaceutical agent met threshold criteria (established for new MCMs) under the federal interagency (PHEMCE) product specific requirements. Filtering based on PHEMCE procurement strategy was done to ensure that the working group conclusions align with broader United States Government efforts to develop nerve agent MCMs according to validated product specific requirements established for de novo development of new antidotes. PHEMCE efforts are targeted to MCM development for civilian population protection, which differ from the Department of Defense requirements with respect to operability requirements (ie, rugged construction, administration without removal of protective ensemble or gas mask, amenable to self- and buddy-administration), but not to toxicity or underlying pathophysiology.
The working group summaries considered a spectrum of antimuscarinic and anticonvulsant agent drug classes. This included repurposed older drugs, widely available medications with FDA indications for non-nerve agent toxicity medical conditions, and effective nerve agent antidotes no longer produced in a compatible formulation. Only currently available, marketed formulations of pharmaceuticals carrying at least one FDA-approved indication are presented in the following.
RESULTS
Standard Therapy
The traditional approach to evaluating a patient exposed to a nerve agent is to determine whether symptoms are present and, if they are, to classify the severity. The recommendations for antidote dosing are based on symptom severity, and most are derived from original work done by the USAMRICD and listed in Table 2.4 A similar civilian classification of nerve agent poisoning severity is presented in training materials, such as the AHLS course.Reference Walter6 Dosing guidance of first-line antidotal drugs, including special population modification, is available on the National Library of Medicine’s Chemical Hazards Emergency Medical Management website.7
Table 2 Standard Classification and Treatment Strategies for Nerve Agents*

1 2.1 mg of atropine and 600 mg of pralidoxime in an autoinjector (Duodote™).
2 2 mg of atropine (Atropen™).
AI=autoinjector; IM=intramuscular.
*Information combined and adapted from the United States Army Medical Research Institute of Chemical Defense (USAMRICD) Medical Aspects of Chemical and Biological Warfare, Volume 3 Table 5.7, and Advanced HazMat Life Support (AHLS), Chemoterrorism: Nerve Agents.” Box 23-1 and 23-2. Mark I Dosing vs. Clinical Severity of Poisoning for Nerve Agent. 8, Reference Thomas9
Public Health Emergency Medical Countermeasures Enterprise (PHEMCE) Recommendations
The working group consensus follows the Department of Health and Human Services PHEMCE Product Specific Requirements for new MCMs for nerve agent poisoning. The working group also agreed with repurposing of available drug formulations as nerve agent MCMs, using novel routes of administration, in line with the 2007 and 2011 PHEMCE requirements and procurement strategy.
The PHEMCE requirement document contains product-specific threshold criteria for new antimuscarinic MCMs:
(1) Centrally acting antimuscarinic agents, which more easily cross the blood-brain barrier than atropine
(2) Inhaled antimuscarinic (eg, dry powdered inhaled atropine) for delivery directly to the lungs to better treat both bronchorrhea and bronchoconstriction
(3) Effective antimuscarinics with a longer half-life
In summary, PHEMCE MCM strategy includes (a) new formulations of existing MCMs (parenteral atropine, parenteral diazepam), which are more easily administered or more readily absorbed; and (b) effective MCMs that can be used by untrained persons at risk, via intranasal or inhaled routes, and by first responders dealing with large numbers of exposed individuals. Table 3 describes the key advantages of (a) the contingency routes of administration and (b) the contingency pharmaceutical agents discussed in detail in the following sections.
Table 3 Advantages of Contingency Medical Countermeasures for NA Poisoning
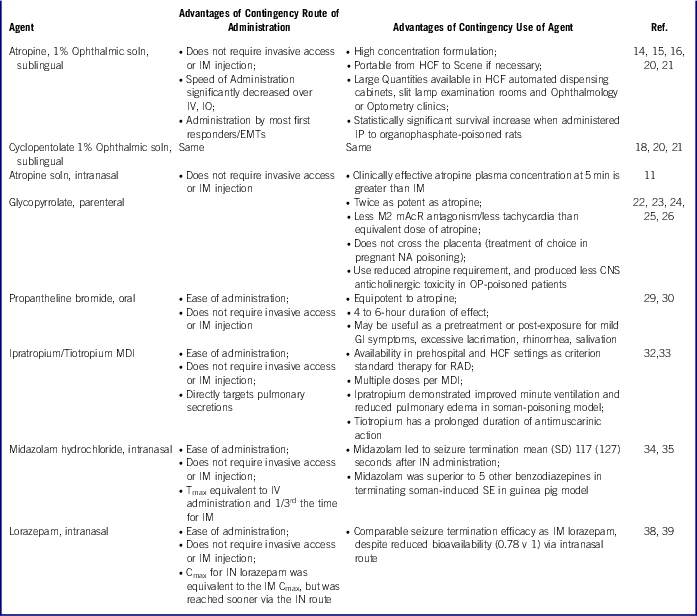
IN – intranasal; IO – intraosseous; IM – intramuscular; IV – intravenous; IP – intraperitoneal; CNS – central nervous system; HCF – healthcare facility; MDI – metered-dose inhaler; SE – status epilepticus; SD – standard deviation; GI – gastrointestinal; RAD – reactive airways disease (asthma); Cmax – peak plasma concentration; Tmax – time post-administration to reach peak plasma concentration; mAchR – muscarinic acetylcholine receptor (where M# denotes receptor subtype/tissue distribution)
Physiology of the Intranasal Route of Administration
The intranasal route of administration has several advantages over other routes, but also has limitations. Intranasal administration enables direct diffusion into the central nervous system through the olfactory epithelium, thus bypassing the blood-brain barrier. In addition, the major blood supply to the largest absorptive area of the nasal mucosa (behind the middle turbinate) is the middle turbinate artery, whose arteriolar anastomoses allow rapid systemic absorption and bypass of first-pass metabolism by the liver.Reference Fraunfelder10 Clinical experience demonstrates that excess doses of ocular solutions, such as eye drops, may pass through the nasolacrimal duct and result in measurable plasma concentrations and systemic effects.Reference Kaila, Korte and Saari11, Reference Shiuey and Eisenberg12 The volume-to-absorptive surface capacity of the nares is a maximum of 1 mL/naris, even when using effective intranasal fine aerosol delivery devices; however, the optimal volume is 0.6 mL/naris.Reference Corrigan, Wilson and Hampton13 Thus, higher concentration preparations are required. Limitations to intranasal administration are nasal trauma, excessive mucous plugging, and blood in the nares, which may restrict absorption.
Options for Antimuscarinic Drugs
Ophthalmic Atropine Solution Administered Sublingual
Atropine sulfate ophthalmic solution, United States Pharmacopeia (USP) 1%, is an extremely potent formulation: each milliliter contains 10 mg of atropine sulfate. Investigators injected 2 mg/0.1 mL of atropine (2%) sublingually in 6 volunteers. Within 10 minutes, the sublingual administration resulted in a greater serum concentration (14 ng/mL) than the therapeutic peak of 6 to 8 ng/mL obtained with a 2-mg intramuscular dose at 30 minutes.Reference Rajpal14 That study does not directly inform our intended use of high concentration ophthalmic atropine preparations for sublingual administration by instillation of drops, nor do we advocate sublingual injection of atropine. However, the study highlights the rapid absorption and improved kinetics from the sublingual space compared with intramuscular administration into the vastus lateralis muscle of the thigh.
In practice, sublingual atropine drops have been used effectively in children (n=23) with severe sialorrhea.Reference Norderyd, Graf and Marcusson15 One drop of 1% atropine ophthalmic (10 mg/mL or approximately 1 mg) was administered sublingually once a day for 4 weeks, followed by twice a day for 4 weeks for palliation of drooling to improve perioral skin integrity and hygiene. Subjective improvement in drooling was reported by parents/caregivers on a visual analog scale; unstimulated saliva production in milliliters per minute (adsorbed to intraoral cotton balls that were weighed) significantly (P=0.032) decreased from baseline to the end of the study. The same approach has been used in cases of severe sialorrhea due to clozapine use.Reference Comley, Galletly and Ash16
Intranasal atropine administration may be an alternative route if an effective delivery device is used with a sufficiently concentrated preparation. Rajpal et alReference Rajpal14 administered 6 mg of atropine (0.6 mL) and 0.5% chitosan to 6 healthy supine adults by means of a nasal catheter to reach the posterior nasopharynx. The viscosity of the mixture was 14.465+0.17 pascal seconds, and clinically relevant blood concentrations of 8 ng/mL were reached in 5 minutes with a maximum concentration (Cmax) of 49 ng/mL at 30 min. Chitosan is a bioadhesive polymer that improves mucosal absorption. In a parallel in vitro model of absorption using rat large intestinal mucosa, the authors showed their formulation’s cumulative drug release to be 0.684 mg/cm2 over 90 minutes, two-thirds (0.4 mg) of which occurred in the first 30–35 minutes before a plateau was reached. Based on these data, the authors estimated the bioavailability of atropine/0.5% chitosan to be 0.40 with an improved time to therapeutic blood concentration and higher Cmax than that with intramuscular administration.17
Also informative to the effectiveness of absorption from the nasal cavity are several case reports of iatrogenic systemic absorption of eye drops and one intraocular pharmacokinetic study documenting systemic absorption.Reference Kaila, Korte and Saari11, Reference Shiuey and Eisenberg12 In 6 volunteers, 30 µL of 1% atropine (0.3 mg) administered as an intraocular drop had a mean bioavailability of 0.63, and a mean Cmax of 288 pg/mL at a mean time to maximum concentration (Tmax) of 27 min.Reference Kaila, Korte and Saari11 Absorption from the nasal mucosa after transiting the nasolacrimal duct was demonstrated in this study by Kaila et al,Reference Kaila, Korte and Saari11 but volume limitations of the canthal space resulted in a delay in reaching Tmax. The use of dilating eye drops in the outpatient setting for sustained miosis has been described in a human trial; 6 healthy volunteers exposed to sarin vapor (0.5 mg/m3 for 30 min) received 60 ul of 1% cyclopentolate intraocular.Reference Moylan-Jones and Thomas18 The authors did not note improvement in visual acuity, refraction, or pupil diameter, but concluded that full dark adaptation (preservation of night vision – a military consideration) or distressing ocular symptoms (pain) might be indications. In a real-world incident, miosis refractory to systemic antimuscarinic therapy is a significant impairment after nerve agent exposure, and mydriatic eye drops are effective therapy. Kato et alReference Kato and Hamanaka19 treated 96 patients for prolonged miosis with 0.5% ophthalmic tropicamide gtt for 3 to 21 days after the 1995 Tokyo subway sarin attack.
Ophthalmic Cyclopentolate Solution Administered Sublingual
Cyclopentolate 1% (1 mL=10 mg) is less potent than atropine but is centrally acting. One percent atropine ophthalmic preparations has been studied for pharmacokinetic profiles via the sublingual and intranasal routes, and 2 studies have examined 3 ophthalmic formulations of various potencies for efficacy against organophosphate pesticide-poisoned rats. Bryant et alReference Bryant, Rhee, Thompson and Aks20 treated 40 rats (n=10 per group saline control, 0.3 mL; atropine sulfate solution, 10 mg/kg of body weight; 1% ophthalmic atropine, 10 mg/kg of body weight; 1% homatropine, 20 mg/kg of body weight) with intraperitoneal drug 10 minutes before an LD50 dose of dichlorvos (10 mg/kg subcutaneous). Survival at 120 minutes (study end point) was 10/10 for atropine sulfate solution, 10/10 with 1% ophthalmic atropine preparation, 9/10 for homatropine, and 2/10 for the control group.Reference Bryant, Rhee, Thompson and Aks20 A subsequent study by the same authors (2009) compared atropine 10 mg/kg, 1% tropicamide 20 mg/kg, and 1% cyclopentolate 20 mg/kg administered intraperitoneally 5 minutes before 15 mg/kg of dichlorvos was given subcutaneously. Survival was 90% in each treatment group (atropine, tropicamide, cyclopentolate) and 10% in controls. In addition to the comparative efficacy results, the authors emphasize the practicality of using high concentration ophthalmic antimuscarinic preparations as a high-capacity contingency: a 15-mL bottle of 1% concentration contains 150 mg of antimuscarinic agent. In their emergency department, the authors identified 125 mg of parenteral atropine in drug-dispensing cabinets and “crash” carts, and 40,000 mg of antimuscarinic agent collectively in ophthalmology preparations.Reference Bryant, Rhee and Thompson21
Parenteral Glycopyrrolate
Glycopyrrolate is 2–3 times as potent as atropine in reducing oral and respiratory secretions.Reference Mirakur, Jones and Dundee22, Reference Gomez, Bellido, Sanchez and de la Cuesta23, Reference Robenshtok, Luria, Tashema and Hourvitz24 Glycopyrrolate is considered a non-selective parenteral muscarinic acetylcholine receptor antagonist used to dry secretions during operative procedures. However, glycopyrrolate may have a lower affinity at muscarinic-2 receptors and induce less tachycardia than atropine.Reference Gomez, Bellido, Sanchez and de la Cuesta23 Glycopyrrolate does not cross the placenta and may be the antimuscarinic of choice in late-term pregnancy for organophosphate or nerve agent intoxication.Reference Robenshtok, Luria, Tashema and Hourvitz24 There are 2 unblinded studies showing comparative efficacy of equipotent doses of atropine and glycopyrrolate in organophosphate poisoning.Reference Bardin and Van Eeden25, Reference Arendse and Irusen26 In 44 human pesticide poisoning cases randomized to either drug, there was no difference in outcome, intubation, or length of stay.Reference Bardin and Van Eeden25 Arendse et alReference Arendse and Irusen26 treated 53 human cases of organophosphate poisoning (2 fatal) with a combination of atropine and an equipotent dose of glycopyrrolate; the authors demonstrated a significant reduction in the incidence (12.2%) of central antimuscarinic toxicity when using glycopyrrolate versus their historical hospital experience treating organophosphate poisoning with atropine alone. Ali-Melkkila et al17, Reference Ali-Melkkila, Kaila and Kanto27 studied the pharmacokinetics of intramuscular (deltoid) glycopyrrolate in 6 ocular surgery patients: after 0.008 mg/kg intramuscularly, the Tmax was 27.48±6.12 minutes with a predictable anti-secretory response; the Cmax was 3.47±1.48 ug/L. The study included a third arm (n=6) dosed with 4 mg po; the absorption was so variable and the onset of clinical effects so unpredictable that the gastrointestinal route of administration is a poor pharmacokinetic/pharmacodynamic-based choice for administration.Reference Ali-Melkkila, Kaila and Kanto27 Kaila et alReference Kaila, Ali-Melkkila, Lisalo and Kanto28 dosed 3 gynecological surgery patients receiving spinal and epidural anesthesia with 0.008 mg/kg glycopyrrolate intramuscularly (deltoid); mean Tmax=13.3 (10–15 minutes) and Cmax=14.26 (12.02–16.97) µg/L. Cerebrospinal fluid concentrations were not detected with an assay level of detection of 140 ng/L.Reference Kaila, Ali-Melkkila, Lisalo and Kanto28
Oral Propantheline
Propantheline bromide has a 1 to 1.25 times higher binding affinity than atropine at the muscarinic receptor and varies slightly by receptor subtype (M1-M4).Reference Huang, Buchwald and Browne29 Propantheline has FDA indications for the treatment of peptic ulcer disease and hyperhidrosis; however, a parenteral formulation of propantheline is no longer produced. The absorption half-life after oral dosing is 8–30 minutes, Cmax was 29 ng/mL, and the Tmax was 3 hours (mean; n=6 healthy volunteers) after 30 mg po with a duration of effect of 4–6 hours.30 Propantheline has not been studied as an oral MCM for mild nerve agent poisoning. Supplies of propantheline in a pharmacy or community are likely to be limited because of its infrequent use. Its pharmacokinetics after oral dosing might make a dose of propantheline an alternative treatment for patients with only mild symptoms (eg, sialorrhea, rhinorrhea, lacrimation, gastrointestinal cramping due to enhanced peristalsis), who might otherwise be treated with a single 2-mg dose of atropine, while they are observed for symptom progression.
Inhaled Ipratropium
The 200 inhalation unit dose of ipratropium bromide has a net weight of 12.9 g. After priming, each actuation of the inhaler delivers 21 µg of ipratropium bromide from the valve in 56 mg of solution and delivers 17 µg of ipratropium bromide from the mouthpiece.31 Ipratropium acts as an antimuscarinic at the neuromuscular junctions in the lung. The half-life of elimination is about 2 hours after inhalation. In 1 study, soman-exposed rats had improved minute ventilation and reduced lung edema after 21 µg of ipratropium, 120 µg of albuterol, 80 µg of budesonide, and 4.5 µg of formoterol administered 10 minutes after exposure; however, no effect on lethality was shown.Reference Perkins, Wong and Rodriguez32
Inhaled Tiotropium
Tiotropium bromide has 23 times the potency of ipratropium for bronchodilation.Reference Barnes33 Antimuscarinic blockade half-life is 540 minutes versus 81 minutes for ipratropium. Each inhaler actuation contains a dry powder consisting of 18 µg tiotropium (equivalent to 22.5 µg of tiotropium bromide monohydrate) blended with lactose monohydrate (which may contain milk proteins and therefore a risk for lactose-intolerant persons).Reference Barnes33
Options for Anticonvulsant Drugs
When evaluating treatment options for the anticonvulsant category, the working group agreed on 4 recommendations summarized in Table 4. The recommendations meet 3 PHEMCE criteria for anticonvulsant nerve agent MCM development, namely (a) improved pharmacokinetic profiles, (b) routes of rapid administration that can be used by more first responders of different skill levels, and (c) comparable efficacy for nerve agent toxicity and clinical pathophysiology (eg, seizures).
Table 4 DHS Working Group Recommendations for the Treatment of Nerve Agent-induced Convulsions

Parenteral Midazolam
Midazolam is a lipid-soluble benzodiazepine that works as an agonist at the GABA-A receptor and possesses anticonvulsant and sedative effects, as documented by pharmacokinetic data presented later.Reference Wermeling, Record and Kelly34 Traditional routes of administration for midazolam are intravenous and intramuscular, and studies have demonstrated absorption and efficacy of intranasal administration. The data we present focus on the pharmacokinetics of anticonvulsant activity where applicable for the intramuscular and intranasal routes.
In a study by Wermeling et alReference Wermeling, Record and Kelly34, intranasal midazolam was compared with intramuscular and intravenous formulations. The study used an open-label, 3-way crossover (separated by 1 week) design, comparing a 5-mg intravenous infusion over 15 minutes with 5 mg given intramuscularly and 5 mg given via the intranasal route. The median time to Tmax was 10.3 min with intranasal administration, 12.4 minutes with intravenous, and 29.2 minutes with intramuscular. The Cmax was 167 ng/mL with intravenous administration, 80 ng/mL with intranasal, and 59 ng/mL with intramuscular. Thus, although Tmax was reached sooner with the drug given via the intranasal route, the intravenous route yielded a higher Cmax. Tmax and Cmax were higher for the intravenous and intranasal formulations than for the intramuscular formulation. Bioavailability was 93% for intramuscular and 72% for intranasal administration compared with an assumed 100% for intravenous administration.Reference Wermeling, Record and Kelly34 Although midazolam given via the intravenous route took longer to reach Tmax than when given via the intranasal route, the concentration of midazolam given by either route was essentially the same at 5 minutes.Reference Wermeling, Record and Kelly34
Another study by Bhattacharyya et alReference Bhattacharyya, Kalra and Gulati35 compared the efficacy of intranasal midazolam with rectal diazepam for the prevention of seizures. In their study, in 46 children who had a total of 188 seizure episodes, 0.3 mg/kg of intranasal midazolam was superior to 0.2 mg/kg of rectal diazepam in terminating seizures within 10 minutes of drug administration, and the time to seizure cessation was 117±127 seconds for intranasal midazolam versus 178±179 seconds for rectal administration (P=0.005).Reference Bhattacharyya, Kalra and Gulati35
The pivotal trial that investigated the efficacy of intramuscular midazolam was that of Silbergleit et al,Reference Silbergleit, Durkalski and Lowenstein36 in which intramuscular midazolam was compared with intravenous lorazepam in the treatment of prehospital status epilepticus in adults and children. All patients weighing over 40 kg were randomized to either 10 mg intramuscular midazolam followed by a placebo infusion, or an intramuscular placebo injection followed by a 4-mg lorazepam infusion; patients 13–40 kg were given 5 mg of midazolam or 2 mg of lorazepam as previously. The study found that 73.4% of the midazolam group arrived at the emergency department without evidence of ongoing seizure compared with 63.4% in the lorazepam group. The median time to treatment was 1.2 minutes for the intramuscular midazolam group versus 4.8 minutes in the lorazepam group. Rates of endotracheal intubation and adverse effects were similar in the 2 groups.Reference Silbergleit, Durkalski and Lowenstein36
McDonough et alReference McDonough, McMonagle and Copeland37 compared 6 separate benzodiazepines in guinea pigs with status epilepticus induced by the nerve agent, soman. The drugs were given at the 5- and 40-minute mark. All benzodiazepines terminated convulsions, but midazolam was the most potent and rapid acting in terminating seizures at both times.Reference McDonough, McMonagle and Copeland37
Intranasal Lorazepam
Lorazepam, like midazolam, is a lipid-soluble benzodiazepine that is an agonist at the GABA-A receptor. Traditional routes of administration are intravenous and intramuscular, but reports support its use via the intranasal route. (Even rectal administration has been evaluated in human subjects; however, slow absorption has limited the drug’s usefulness when administered rectally.Reference Graves, Kriel and Jones-Saete38)
In a study by Wermeling et alReference Wermeling, Miller and Archer39, 11 healthy volunteers underwent an open, 3-arm crossover study in which each volunteer received 2 mg of lorazepam via intravenous, intramuscular, or intranasal routes after adequate washout periods between each administration. The median Tmax for intravenous lorazepam was 6 (range 4.9–60) minutes; for intranasal, 30 (range 15–120) minutes; and for intramuscular, 180 (range 30–480) minutes. The Cmax was 47.6 ng/mL for intravenous lorazepam, 22.6 ng/mL for intramuscular lorazepam, and 21.4 ng/mL for intranasal lorazepam. Bioavailability was 100% for intramuscular lorazepam and 78% for intranasal lorazepam, compared with an assumed 100% bioavailability for intravenous administration.Reference Wermeling, Miller and Archer39 Thus, intranasal lorazepam yielded a Cmax similar to intramuscular lorazepam, but it reached the Cmax significantly earlier. However, because of less bioavailability with the intranasal route, the area under the curve still favored the intramuscular route.Reference Wermeling, Miller and Archer39 Therefore, the clinician must weigh the importance of achieving rapid Cmax versus total drug absorbed. The authors did not find a trial that directly compared intramuscular lorazepam with intranasal midazolam.
Parenteral Diazepam
Diazepam also is a benzodiazepine that is active at the GABA-A receptor. As mentioned in the Working Group consensus (see Table 4), all parenteral benzodiazepines can terminate convulsions caused by a nerve agent. Historically, when AIs of benzodiazepines were introduced for the treatment of nerve-agent exposure, the benzodiazepine that was FDA-approved for the treatment of status epilepticus was diazepam, given intravenously. It followed that diazepam could be packaged in an AI for the treatment of nerve agent-induced convulsions.Reference McDonough40 However, intramuscular diazepam is absorbed slowly, incompletely, and erratically from the site of administration.Reference Hung, Dyck and Varvel41, Reference Divoll, Greenblatt, Ochs and Shader42 In a study of healthy volunteers, 5 mg of midazolam or 10 mg of diazepam were injected into opposite deltoid muscles. The Cmax was 100.5±21 ng/mL for midazolam and 199±89.3 ng/mL for diazepam. The corresponding Tmax was 17.5±6.5 minutes (midazolam) versus 33.8±7.5 minutes (diazepam).Reference Hung, Dyck and Varvel41 Intramuscular absorption was significantly more variable for diazepam than for midazolam.Reference Divoll, Greenblatt, Ochs and Shader42 Diazepam can be administered rectally, but the plateau state was reached more slowly than with midazolam given via the intramuscular route.Reference Rey, Tréluyer and Pons43
As stated previously, intramuscular midazolam terminated soman-induced convulsions more rapidly than did other benzodiazepines, including diazepam, in guinea pigs.Reference McDonough, McMonagle and Copeland37 This observation has led to a preference of intramuscular midazolam over intramuscular diazepam for the treatment of nerve-agent convulsions, despite the historical use of AI diazepam. Although diazepam AIs remain the conventional NA anticonvulsant stockpiled for the military and in national stockpile caches, additional pharmacokinetic studies support the opinion that intramuscular diazepam is not necessarily the foremost option for treatment of nerve agent-induced convulsions if intramuscular midazolam and intramuscular lorazepam are also available.
CONCLUSIONS
Nerve agent MCM stockpiles at the local community level may not be adequate for treating mass human exposure after a terrorist attack or an accidental agricultural or industrial release of organophosphate pesticides. A DHS subject-matter expert working group, with the collaboration of federal interagency partners, identified alternative pharmaceutical MCMs for the treatment of nerve agent toxicity when FDA-approved drugs deployed on AI platforms (first-line antidotes) are unavailable. The medications reviewed here align with governmental strategies for de novo nerve agent MCMs research and development, which favor ease of use by a range of trained first responders, speed and ease of administration, favorable pharmacokinetic profiles allowing for less re-dosing, and reduced drug use per patient. To address concerns about current stockpile sustainability and near-term sourcing challenges of first-line (AI-based platforms) MCMs, the working group limited its consideration to pharmaceuticals with FDA approval for at least 1 indication, those currently marketed in the United States, and those with appropriate formulation without modification for PHEMCE-preferred routes of administration.




