


I. INTRODUCTION
Because of severe side effects, selectivity problems and resistance development potential of drugs used in cancer chemotherapy, the discovery of new drugs in this area is still a major topic. In recent years, combined anticancer therapies or multi-acting drugs are clinically preferred over the traditional cytotoxic treatment, with the aim of avoiding resistance and toxic side effects. The arrangement of multi-acting targets can be carried out either by the combination of several drugs with different mechanisms or by the usage of a single chemical compound capable of regulating several targets of a disease with multiple factors. The emergence of this information has greatly increased the interest in the discovery of new compounds addressing multiple biological targets.
There are many studies reporting the anticancer and cancer chemopreventive effects of conventional non-steroidal analgesic anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors (Amir and Agarwal., Reference Amir and Agarwal2005; Guadagni et al., Reference Guadagni, Ferroni, Palmirotta, Del Monte, Formica and Roselli2007; Cuzick et al., Reference Cuzick, Otto, Baron, Brown, Burn, Greenwald, Jankowski, La Vecchia, Meyskens, Senn and Thun2009; Gupta et al., Reference Gupta, Sung, Prasad, Webb and Aggarwal2013). It is also well documented that 1,2,4-triazole is one of the important core fragments that is incorporated in many anticancer agents. In recent years, a number of condensed 1,2,4-triazole derivatives such as triazolothiadiazole, triazolopyridazine, triazolotriazine, and triazolothiadiazine have attracted attention because of their cytotoxic effects (Zhang et al., Reference Zhang, Vojkovsky, Huang, Liang, Do, Koenig and Cui2005; Lauffer et al., Reference Lauffer, Aronov, Li, Deininger, Mcginty, Stamos, Come and Stewart2007, Reference Lauffer, Li and Mcginty2010; Lesyk et al., Reference Lesyk, Vladzimirska, Holota, Zaprutko and Gzella2007; Albrecht et al., Reference Albrecht, Harmange, Bauer, Berry, Bode, Boezio, Chen, Choquette, Dussault, Fridrich, Hirai, Hoffman, Larrow, Kaplan-Lefko, Lin, Lohman, Long, Moriguchi, O'Connor, Potashman, Reese, Rex, Siegmund, Shah, Shimanovich, Springer, Teffera, Yang, Zhang and Bellon2008; Bhat et al., Reference Bhat, Poojary, Prasad, Naik and Holla2009; Boezio et al., Reference Boezio, Berry, Albrecht, Bauer, Bellon, Bode, Chen, Choquette, Dussault, Fang, Hirai, Kaplan-Lefko, Larrow, Lin, Lohman, Potashman, Qu, Rex, Santostefano, Shah, Shimanovich, Springer, Teffera, Yang, Zhang and Hermange2009; Ibrahim, Reference Ibrahim2009; Khan et al., Reference Khan, Zaib, Ibrar, Rama, Simpson and Iqbal2014b). Developing new compounds by combining several active groups into a single molecule can enhance the efficiency of the compounds compared with single-target drugs because of modulate multiple cellular pathways. In the light of this knowledge, in an earlier study, novel condensed triazol derivatives with the structure of triazolothiadiazine having structural motifs of NSAIDs were synthesized and investigated for their bioactivity against epithelial cancer cells (Aytaç et al., Reference Aytaç, Durmaz, Houston, Çetin-Atalay and Tozkoparan2016). The results we obtained in that study showed that these novel hybrid compounds can be good candidates for the treatment of epithelial cancers particularly liver cancer. The title compound is one of the members of this series having cytotoxic activity.
The authors present here the crystal structure of 3-[1-(6-methoxy-2-naphtyl)ethyl]-6-(2,4-dichlorophenyl)-7H-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine (Figure 1) determined by synchrotron powder X-ray diffraction (XRD) analysis.
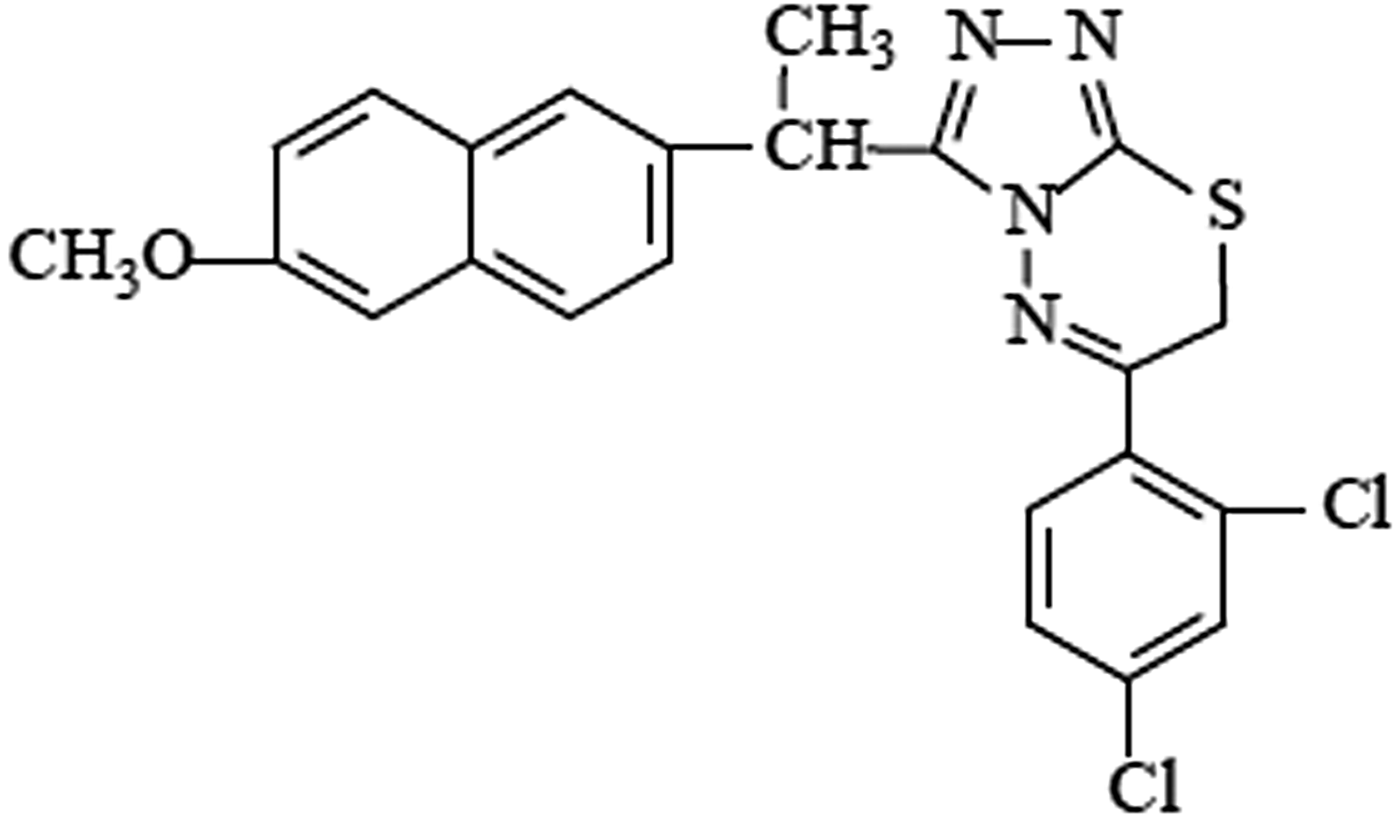
Figure 1. The structural formula of the title compound in the two-dimensional structure form.
II. EXPERIMENTAL
A. Synthesis and crystallization
The synthesis of the title compound was carried out simply by refluxing equimolar amounts of 4-amino-3-[1-(6-methoxy-2-naphtyl)ethyl]-1,2,4-triazole-5-thione and 2,4-dichlorophenacyl chloride in ethanol. The required starting compound was obtained by the reaction of naproxen with thiocarbohydrazide by employing previously reported procedure (Aytaç et al., Reference Aytaç, Durmaz, Houston, Çetin-Atalay and Tozkoparan2016). The chemical composition of the compound was confirmed by spectral and elementary analysis, all results were given previously (Aytaç et al., Reference Aytaç, Durmaz, Houston, Çetin-Atalay and Tozkoparan2016).
B. XRD data collection
This compound has a tendency to crystallize as a very fine powder. No crystal of sufficient thickness and quality could be obtained to perform a single-crystal analysis, so the crystal structure determination was done by powder XRD.
First, a Bruker D8 Advance X-ray diffractometer with Mo-anode was used to collect the data. Strong peak overlap was observed in the powder pattern because of the instrumental broadening. The powder pattern was indexed successfully, but structure solution could not be obtained. The peak overlap limits the reliability of intensity information. Then a high-resolution synchrotron measurement of the title compound was done at the beamline 11-BM at the Argonne National Laboratory using the mail-in program. This beamline has a unique multi-analyzer detection assembly, consisting of 12 independent Si (111) crystal analyzers and LaCl3 scintillation detectors. The powder sample was contained in a 0.8 mm diameter kapton capillary and spun at 60 Hz during data collection. Powder XRD data were collected from 0.5 to 50° 2θ range with a step size of 0.001° at room temperature (295.0 K) with a wavelength of 0.459169 Å.
C. Indexing
The synchrotron powder XRD pattern of the title compound was indexed using the program TOPAS 4.2 (Coelho, Reference Coelho2009) giving the following unit cell with monoclinic metric symmetry P21; a = 15.557 Å, b = 8.616 Å, c = 8.566 Å, β = 104.3°, V = 1112.7 Å3 (Gof = 159.04). The synchrotron powder pattern was also indexed with the programs McMaille (Le Bail, Reference Le Bail2004), DICVOL (Boultif and Louer, Reference Boultif and Louer1991), ITO (Visser, Reference Visser1969), and NTREOR (Altomare et al., Reference Altomare, Giacovazzo, Guagliardi, Moliterni, Rizzi and Werner2000) in EXPO (Altomare et al., Reference Altomare, Cuocci, Giacovazzo, Moliterni, Rizzi, Corriero and Falcicchio2013). The similar monoclinic cell was found with a = 15.560 Å, b = 8.617 Å, c = 8.569 Å, β = 104.3°, V = 1113.5 Å3 (M N = 51.3, F N = 576.0) by DICVOL; a = 15.559 Å, b = 8.617 Å, c = 8.567 Å, β = 104.3°, V = 1112.9 Å3 (FOM = 101.7) by ITO; a = 15.554 Å, b = 8.618 Å, c = 8.566 Å, β = 104.3°, V = 1112.6 Å3 (FOM = 391.76) by McMaille; and a = 15.556 Å, b = 8.616 Å, c = 8.568 Å, β = 104.3°, V = 1112.8 Å3 (M 20 = 176) by NTREOR.
D. Structure solution
A rigid body of the molecule in the z-matrix notation was prepared using the Avogadro molecule drawing program (Hanwell et al., Reference Hanwell, Curtis, Lonie, Vandermeersch, Zurek and Hutchison2012) including H atoms and optimized. The H atoms were removed because of their low scattering power and were not added up to the final refinement step. The unit-cell parameters obtained by TOPAS 4.2 and the z-matrix of the molecule were implemented in the DASH program (David et al., Reference David, Shankland, van de Streek, Pidcock, Motherwell and Cole2006) to obtain starting structural model. The diffraction intensities were extracted with the Pawley method and then the simulated annealing was performed. During the simulated annealing calculations, the full pattern was used and the molecule conformation in the unit cell and torsion angles changed randomly. After nearly 10 million movements in the simulated annealing, the best solution (profile χ2 = 5.1360 and intensity χ2 = 223.0303) was gained.
E. Rietveld refinement
The structure solution found by DASH program was introduced in FullProf program (Rodriguez-Carvajal, Reference Rodriguez-Carvajal1993) to perform the Rietveld refinement. The refinement was performed using only the data from 1.2° to 25° 2θ range. The whole profile fitting was done using the LeBail method with 34 points chosen arbitrarily for defining the background by interpolation on the powder pattern. The X-ray powder pattern of the title compound is given in Table I. The particular peak that has the strongest intensity in the entire pattern is assigned of 1000 and other lines are scaled relative to this value. During the refinement, two asymmetry correction parameters with the pseudo-Voigt profile shape function (Thompson et al., Reference Thompson, Cox and Hastings1987) were used, which allow for angle-dependent asymmetry because of axial divergence (Finger et al., Reference Finger, Cox and Jephcoat1994).
Table I. X-ray powder pattern of the title compound [P21, Z = 2], a = 15.55617(10) Å, b = 8.61683(6) Å, c = 8.56690(5) Å, β = 104.32732(41)°, and V = 1112.633(13) Å3.

The particular peak that has the strongest intensity in the entire pattern is assigned of 1000 and other lines are scaled relative to this value. The d-calc values are calculated values from refined lattice parameters. “I obs ” represents integrated intensity values and “M” represents multiplicity.
The refinement without the soft restrains of the bond lengths and angles failed as expected. To limit the number of free parameters, to gain chemically meaningful structure, and to achieve the convergence of the refinement, soft restraints for all bond lengths and angles were used during the refinement. Soft restraints were used and the atom positions were refined in two steps. At the first step, the sigma values, defining the weights of restraints, were set to 0.05° for the bond angles and 0.005 Å for the bond lengths. At the second step, the sigma values were set to 0.1° and 0.01 Å for the bond angles and bond lengths, respectively (Ivashkevich et al., Reference Ivashkevich, Serebryanskaya, Lyakhov and Gaponik2011).
At the beginning of the refinement of atomic positions, the coordinates of the Cl atoms were refined. Then the coordinates of the S and O atoms were refined, respectively. While refining one type of atom position, the other positions of atoms were kept fixed. The atoms of each ring were refined together followed by one ring after another.
The displacement parameters were constrained for each atom type and refined isotropically from the heavy atoms to the light atoms individually. All non-H atoms were located in their calculated positions and a reasonable solution was obtained with R p = 0.06, R wp = 0.076, and χ2 = 1.16 confidence values.
Finally, the hydrogen atoms were placed geometrically with their calculated positions [C−H = 0.96 Å and B iso (H) = 1.5 B iso (C)] using Crystals program (Cooper et al., Reference Cooper, Thomson and Watkin2010). The sigma values of the hydrogen atoms were set to 0.05° and 0.00001 Å for the bond angles and bond lengths, respectively, and were fixed at their calculated atomic positions. At each step, profile and unit-cell parameters were also refined, simultaneously. After the final Rietveld refinement, a good agreement between calculated and experimental powder diffraction patterns was achieved with the confidence values of R p = 0.055, R wp = 0.069, and χ2 = 0.9705 (Figure 2).
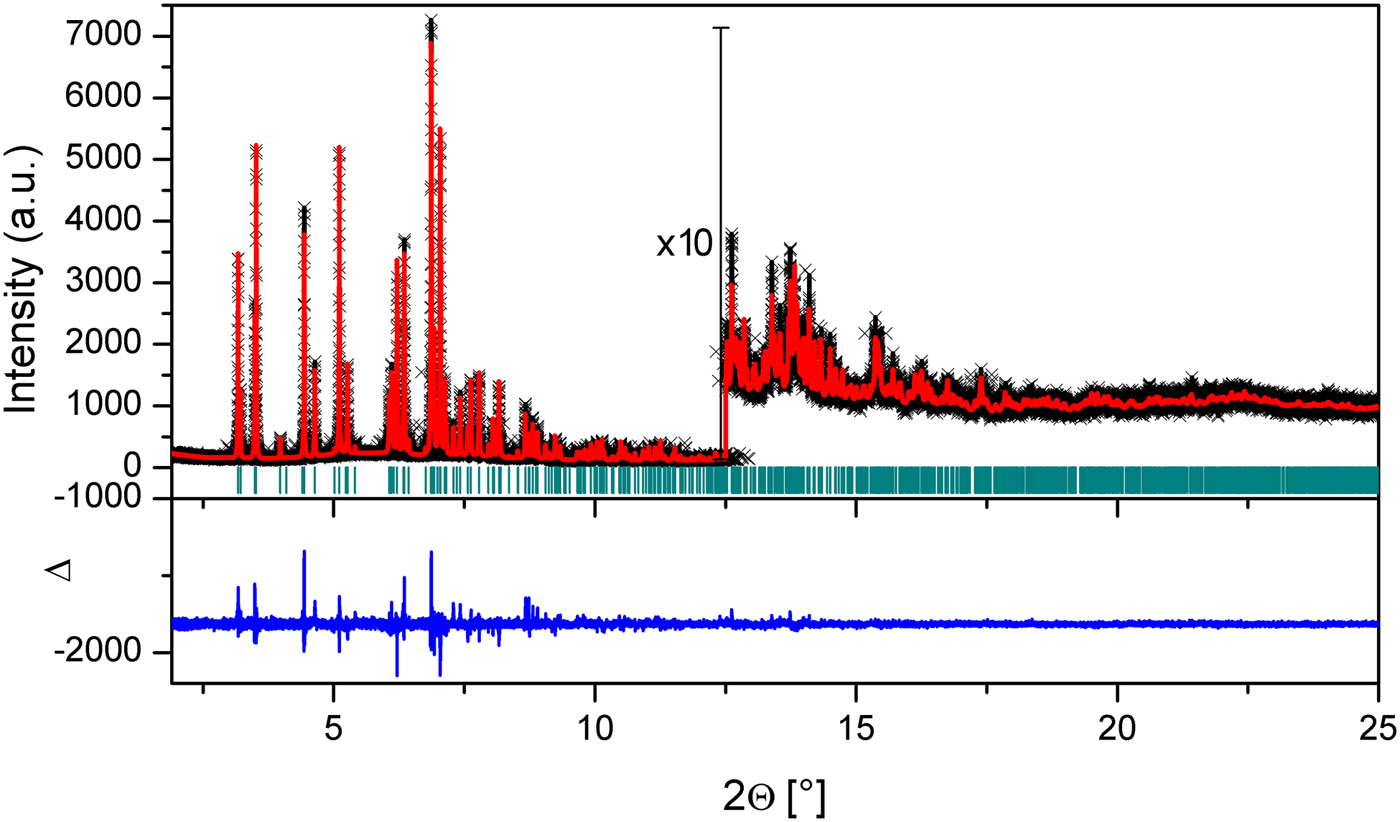
Figure 2. (Color online) Plot of the Rietveld refinement of the title compound. The red dots correspond to the measured powder diffraction pattern and the black line to the calculated one. The difference curve is shown in blue. Green vertical bars indicate Bragg reflections.
III. DISCUSSION
Crystal data, data collection, and structure refinement details are summarized in Table II. A thermal ellipsoid plot of the molecule was prepared using Ortep-3 (Farrugia, Reference Farrugia2012) with the atomic numbering (Figure 3). The probability of the ellipsoid was set to 50%. The selected bond lengths, bond angles, and torsion angles of the title compound are listed in Table III. For calculating molecular interactions and molecular geometry calculations, Platon (Spek, Reference Spek2003), Mercury (Macrae et al., Reference Macrae, Edgington, McCabe, Pidcock, Shields, Taylor, Towler and van de Streek2006), and OLEX2 (Dolomanov et al., Reference Dolomanov, Bourhis, Gildea, Howard and Puschmann2009) programs were used.
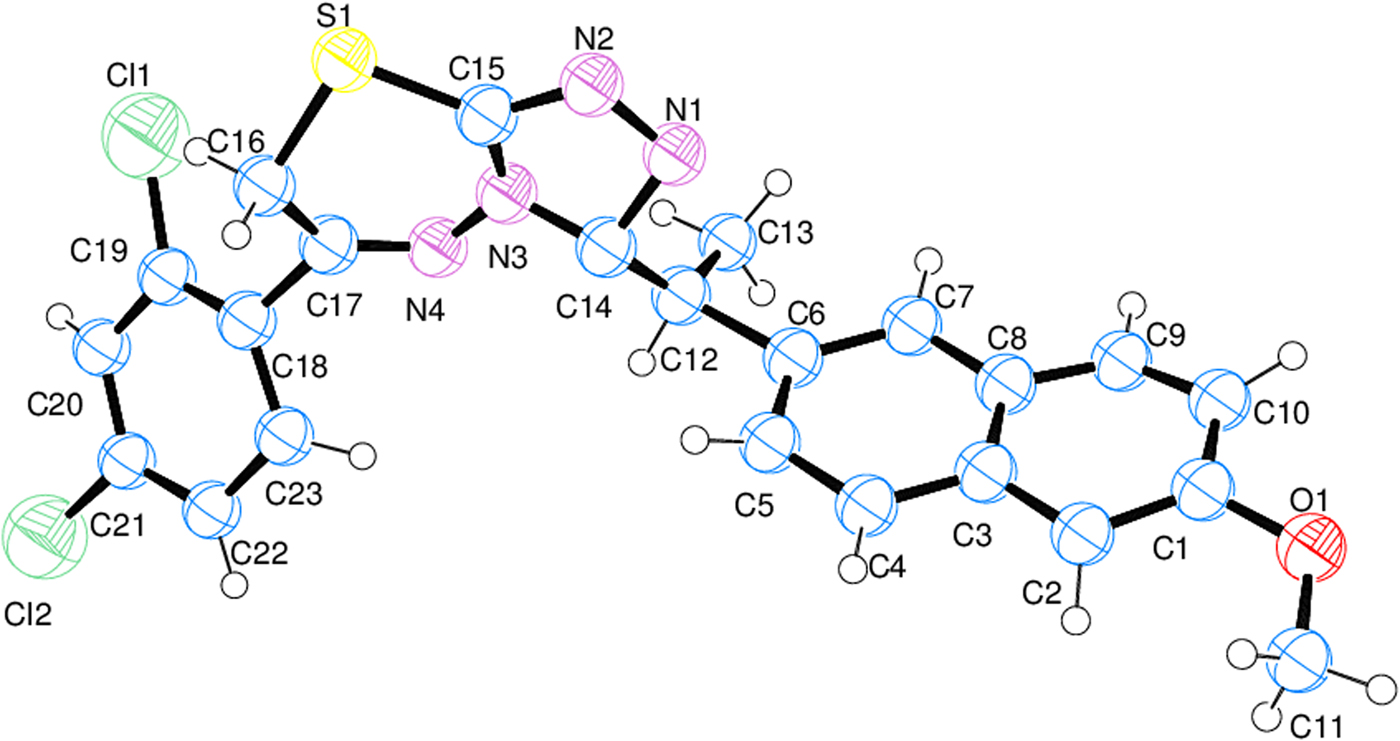
Figure 3. (Color online) Thermal ellipsoid plot of the molecule prepared by Ortep. The thermal ellipsoids are set to 50% probability.
Table II. Crystal data and structure refinement parameters of the title compound.

Table III. Selected geometric parameters (Å, °) for the title compound.

In the title compound, all of the bond lengths and angles in the phenyl rings are in the normal range. In the 1,2,4-triazole ring, the C = N bond lengths are 1.347(13) Å and 1.354(11) Å, which are longer than that found in the 1,2,4-triazole ring-containing structures (Abdel-Aziz et al., Reference Abdel-Aziz, Ng and Tiekink2011; Aytaç et al., Reference Aytaç, Tozkoparan, Kaynak, Aktay, Göktaş and Ünvar2009; Fun et al., Reference Fun, Chantrapromma, Bhat and Abdel-Aziz2012; Kaynak et al., Reference Kaynak, Aytaç and Tozkoparan2010; Khan et al., Reference Khan, Ibrar, Zaib, Ahmad, Furtmann, Hameed, Simpson, Bajorath and Iqbal2014a). The N–N bond length is 1.356(9) Å, which is shorter than the 1,2,4-triazole ring-containing structures cited previously. In the 1,3,4 thiadiazine ring, the bond lengths are compatible with the 1,2,4-triazole ring and the 1,3,4 thiadiazine ring-containing structures (Aytaç et al., Reference Aytaç, Tozkoparan, Kaynak, Aktay, Göktaş and Ünvar2009; Kaynak et al., Reference Kaynak, Aytaç and Tozkoparan2010; Abdel-Aziz et al., Reference Abdel-Aziz, Ng and Tiekink2011; Fun et al., Reference Fun, Chantrapromma, Bhat and Abdel-Aziz2012), except the N–N bond length with 1.325(12) Å. The 1,3,4-thiadiazine ring is not planar and the r.m.s. deviation from planarity is 0.173 Å. S1 atom is displaced from the mean plane C15/N3/N4/C17/C16 by 0.61 Å. The 1,2,4-triazolo ring is planar with r.m.s. deviation 0.027 Å. The phenyl rings are approximately planar with r.m.s. deviation 0.037 Å (C3−C8), 0.068 Å (C18−C23), and 0.056 Å (C1/C2/C3/C8/C9/C10). Atom Cl2 is almost in the same plane with the C18−C23 ring, but the Cl1 atom is placed 0.203 Å below this plane. Structural results show that the C1/C2/C3/C8/C9/C10 ring adopts the envelope conformation having spherical polar set values of Q = 0.138(9) Å, θ = 63(4)°, and φ = 60(4)°.
The dihedral angle between the 1,2,4-triazolo and 1,3,4-thiadiazine rings is 11.1(5)° and between the 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine ring and the 2,4-dichlorophenyl ring is 50.74°. The 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine ring is almost perpendicular to 3-[1-(6-methoxy-2-naphtyl)ethyl] ring with an angle of 86.72°.
The crystal structure of the title compound is stabilized by inter- and intra-molecular hydrogen bonds. The hydrogen-bonding geometry is given in Table IV and is shown in Figure 4. As can be seen from the packing diagram in Figure 5, the molecules are in a head-to-head arrangement. In the crystal structure, there are also strong C−H⋯π and weak intermolecular hydrogen-bonding interactions which link the molecules into a three-dimensional network. The C−H⋯π interactions are C16−H162⋯Cg(C3−C8) and C22−H221⋯Cg(1,2,4-triazole). The distance of H162 to the centroid of the C3−C8 ring is 2.54 Å (symmetry code: −x,1/2 + y,−z) and the centroid angle is 140° (Figure 6). The distance of H221 to the centroid of the 1,2,4-triazole ring is 2.94 Å (symmetry code: x,1 + y, z) and the centroid angle is 118° (Figure 7).
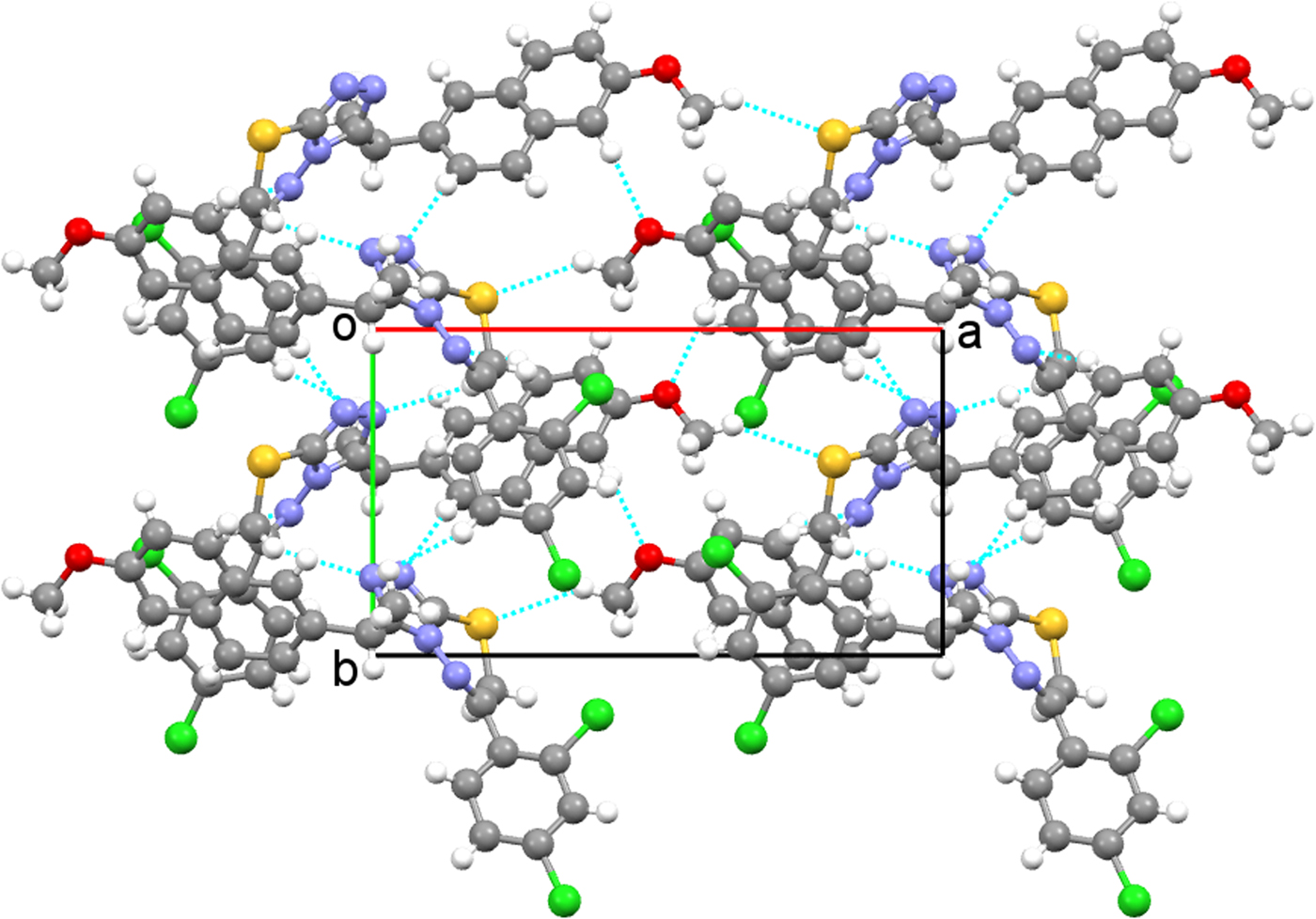
Figure 4. (Color online) Intermolecular hydrogen bonding interactions.

Figure 5. (Color online) Crystal packing of the title compound projected onto ac plane.
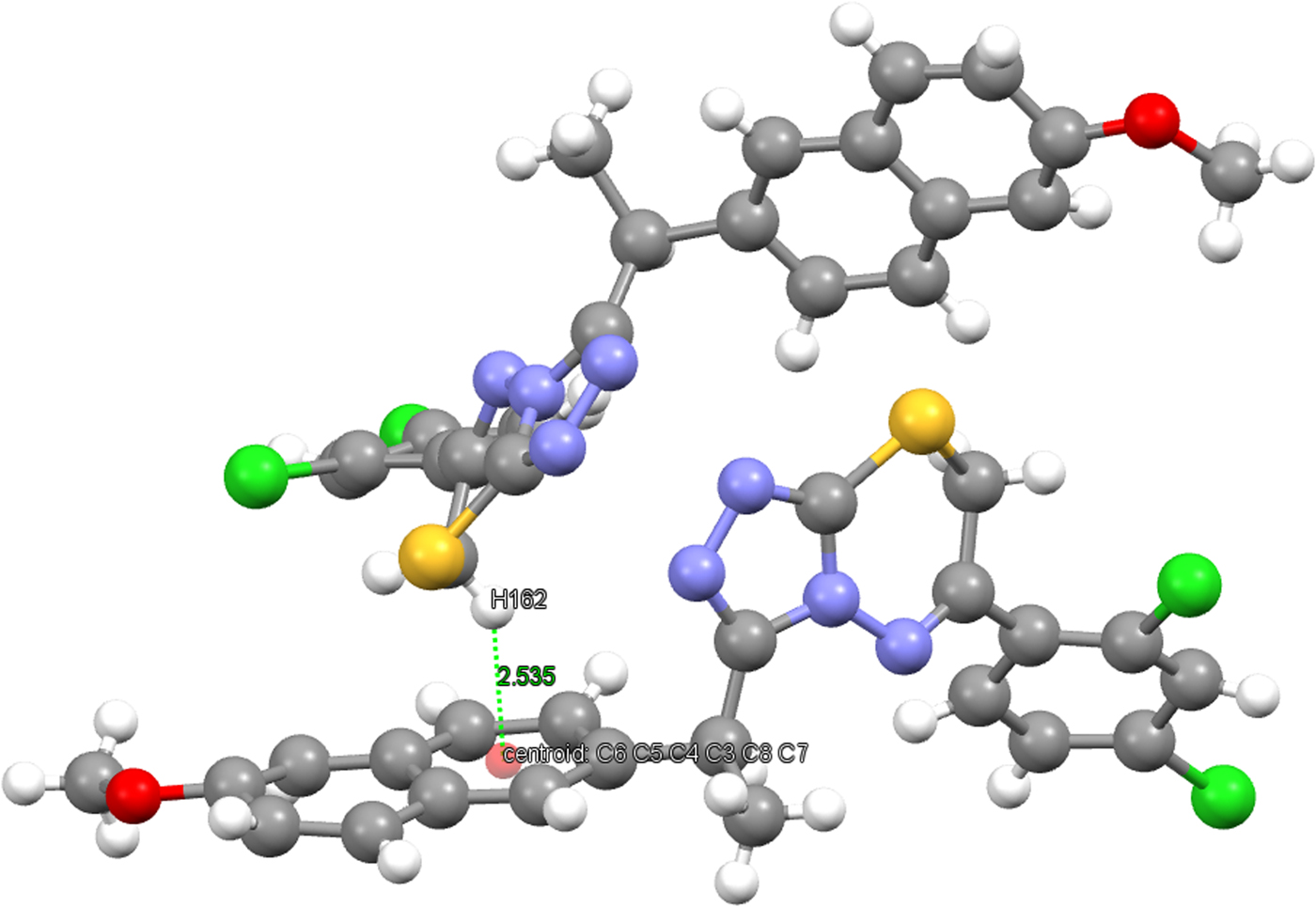
Figure 6. (Color online) C(16)−H(162)⋯Cg(C3−C8) interaction.
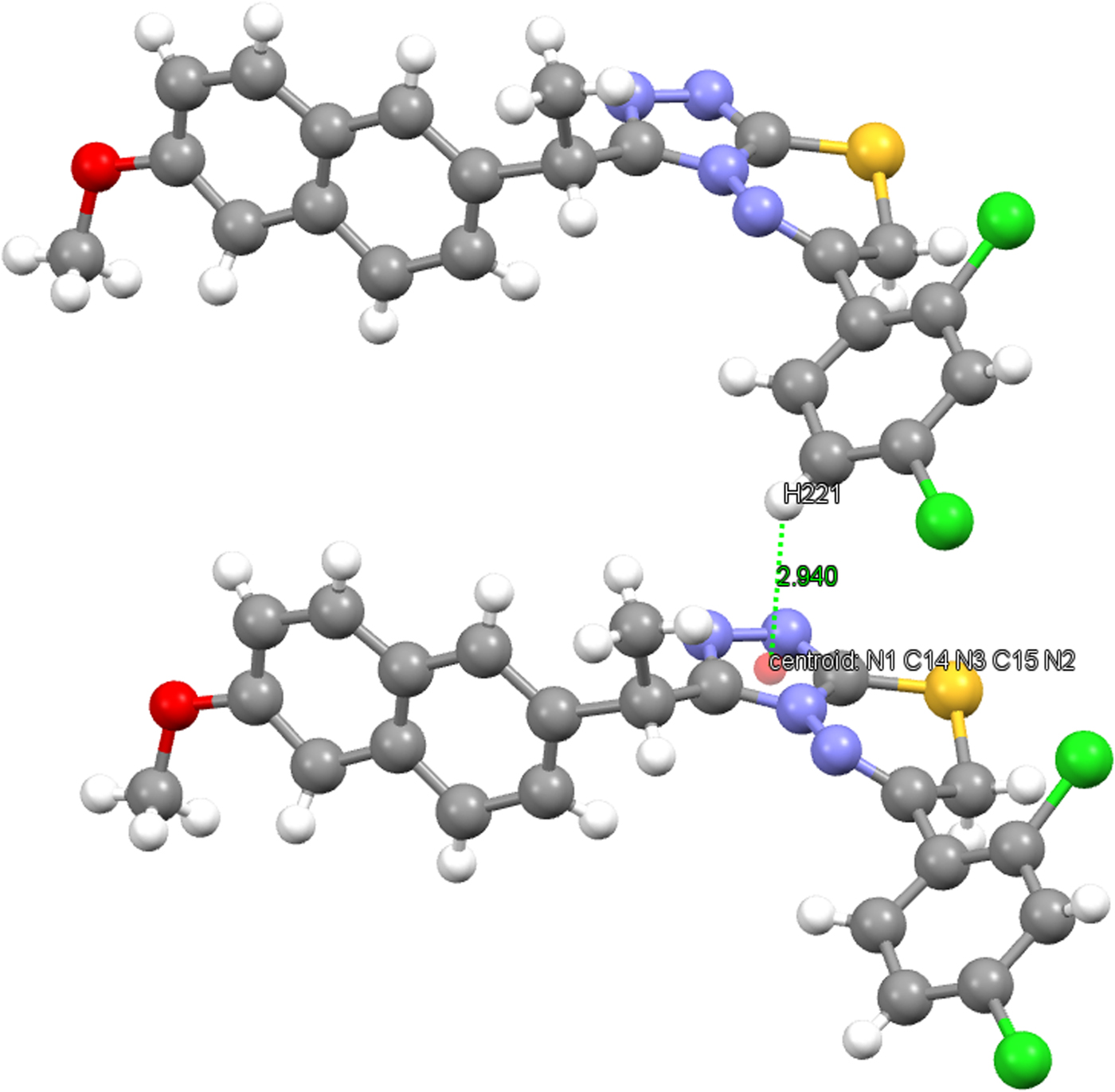
Figure 7. (Color online) C(22)−H(221)⋯Cg(1,2,4-triazole) interaction.
Table IV. Hydrogen-bond geometry (Å, °) for the title compound.

Symmetry codes: (i) −x, y + 1/2, −z; (ii) −x, y−1/2, −z + 1; (iii) x−1, y, z−1.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715617001099.
NOTE
This study has been performed from the PhD thesis of Gülsüm Gündoğdu.
ACKNOWLEDGEMENTS
Financial support to Gülsüm Gündoğdu from The Scientific and Technological Research Council of Turkey (TÜBİTAK) is gratefully acknowledged (2214-A 1-year international research grant m during PhD). She is indebted to Professor Hermann Gies, head of the Ruhr University Bochum, Institute for Geology, Mineralogy and Geophysics, Department of Crystallography, Work Group for Crystal Chemistry, for hosting her in his group during the period of 1 year. She is also grateful to Dr. Bernd Marler, member of the staff working team Gies, for his help in collecting the Laboratory powder XRD data. Use of the Advanced Photon Source at the Argonne National Laboratory was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.













