INTRODUCTION
The Drugs for Neglected Diseases initiative (DNDi) is a patients' needs-driven product development partnership created in 2003 which aims to address the lack of efficacious and safe treatments for neglected diseases. DNDi has adopted a pragmatic virtual R&D model operating in collaboration with partners from pharmaceutical as well as non-pharmaceutical sectors. The organization has a portfolio of research and development projects spanning the kinetoplastid infections (human African trypansomiasis, leishmaniasis and Chagas' disease), paediatric HIV infection, malaria and the filarial infections, onchocerciasis and lymphatic filariasis (www.dndi.org/diseases-projects/portfolio.html). At the research stage, DNDi concentrates its efforts on the establishment of a healthy and diverse pipeline for the filarial infections, human African trypanosomiasis, visceral leishmaniasis and Chagas' diseases, bearing in mind the inevitable attrition rate related to any R&D programme (Ioset and Chang, Reference Ioset and Chang2011). This article will focus on strategies related to the screening of chemical libraries to identify novel lead series against the kinetoplastid parasites, Leishmania donovani (leishmaniasis) and Trypanosoma cruzi (Chagas' disease).
SCREENING ASSAYS
A major consideration for DNDi when developing its early discovery strategy has been the complexity of target validation (Sams-Dodd, Reference Sams-Dodd2005) and subsequent paucity of validated targets essential to the parasite which would be amenable for the development of assays allowing high-throughput screening in the field of neglected diseases (Wyatt et al. Reference Wyatt, Gilbert, Read and Fairlamb2011). Indeed little has so far translated into R&D breakthroughs despite huge expectations following the publication of the complete ‘Tritryp’ (Leishmania major, Trypanosoma brucei and T. cruzi) genome sequences in 2005, revealing that each genome contains 8300–12 000 protein-coding genes, of which around 6500 are common to all 3 genomes. It is fair to say that only a limited number of targets reviewed elsewhere (Renslo and McKerrow, Reference Renslo and McKerrow2006), mostly related to T. brucei sub-species, have so far reached a substantial level of biological characterization and validation in spite of the rapid development of the technologies and methodologies required for their assessment (Frearson et al. Reference Frearson, Wyatt, Gilbert and Fairlamb2007). As far as kinetoplastid diseases are concerned, most of the drugs used in the field act via unclear or unknown mechanisms of action (Castillo et al. Reference Castillo, Dea-Ayuela, Bolas-Fernandez, Rangel and Gonzalez-Rosende2010). From the target-based assays implemented for screening purposes, few were able to deliver progressable candidates (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010) and none have led to new drugs or drug candidates in clinical research. This is partly due to a lack of translation from the target-based activity to whole-cell assays or in vivo activities. This observation is unfortunately not new to the field of anti-infective drug discovery, with similar hopes raised from the first publication of the bacterial genome in 1995, which triggered pharmaceutical companies to move back to antibacterial research and development using genomics-derived, target-based strategies to screen for new classes of drugs with novel modes of action. Despite the evaluation of more than 300 genes, together with 70 high-throughput screening campaigns at Glaxo SmithKline (GSK) over a period of 7 years, there was a very modest yield. A mere 16 campaigns were able to deliver hits and only 5 of these could be turned into leads (Payne et al. Reference Payne, Gwynn, Holmes and Pompliano2007). Lessons learnt from this, and other specific analyses have dramatically influenced antibacterial drug discovery strategies within pharmaceutical companies over the last decade. GSK took the strategic decision to rely as early as possible on in vitro phenotypic assays and in vivo models to characterize the biological activity of the molecules under investigation, together with the use of HTS whole-cell assays for screening. Similarly, Product Development Partnerships involved in drug discovery activities for malaria (MMV), tuberculosis (TB Alliance) and kinetoplastids (DNDi) have opted for medium- to high-throughput screening based on whole-cell assays to evaluate compound libraries accessed from their partners for hit identification. This cell-based screening approach has already been successfully used to identify several novel clinical candidates including thiazolidinones, spiroindolones and diarylquinolines for the treatment of hepatitis C, malaria and tuberculosis respectively, as recently reviewed by Keller et al. (Reference Keller, Shi and Wang2011).
The intracellular amastigotes of L. donovani and T. cruzi in the host cells of infected tissues are the focus of drug discovery efforts. Because of the relative failure of target-based screening in anti-infective drug discovery, since 2008 DNDi has strategically invested in the development and validation of high-content phenotypic screening assays against the intracellular stages of these parasites, in partnership with Institut Pasteur Korea and the University of Dundee. These assays are based on imaging analysis technology that specifically differentiates between infected and non-infected cells via the spatial localization and segmentation of specific descriptors related to the host cell and the parasites. The development of those high-content screening assays against L. donovani (Siqueira-Neto et al. Reference Siqueira-Neto, Moon, Jang, Yang, Lee, Moon, Chatelain, Genovesio, Cechetto and Freitas-Junior2012) and T. cruzi, in combination with an increased 384-well throughput capacity against the extracellular Trypanosoma brucei brucei at the Eskitis Institute for Cell and Molecular Therapies, Griffith University (Sykes and Avery, Reference Sykes and Avery2009), have enabled the screening of medium (10 000–50 000) to large size (50 000–200 000) commercial and proprietary libraries accessed by DNDi. To date, this screening platform has allowed evaluation of more than one million compounds, leading to the identification of numerous novel active series against all three kinetoplastids, many of which have been, or are currently being, progressed in the lead optimization phase. In addition to a dramatic increase in screening throughput against the intracellular amastigote form of the pathogens, high-content assays are also more informative and more specific than colorimetric or fluorimetric assays. As such, it is possible to discriminate between activity resulting from specific pathogen killing versus activity due to host cell damage, or any sub-cellular event possibly indicative of a specific mechanism of action. Such additional image analysis information can be used immediately after screening or archived in anticipation of any subsequent complementary analysis.
SELECTION OF CHEMICAL LIBRARIES: DNDI'S APPROACHES
The introduction of automated high-content screening has led to very significant increases in screening throughput of intracellular Leishmania spp. and T. cruzi amastigotes over the last five years. Nevertheless, compared to the classic throughput achieved in pharmaceutical companies, with a capacity for tens of thousands of screening points per month, it can still only be considered as a medium-throughput screen. Therefore judicious selection and prioritization of chemical libraries is necessary to take advantage of the still limited capacity.
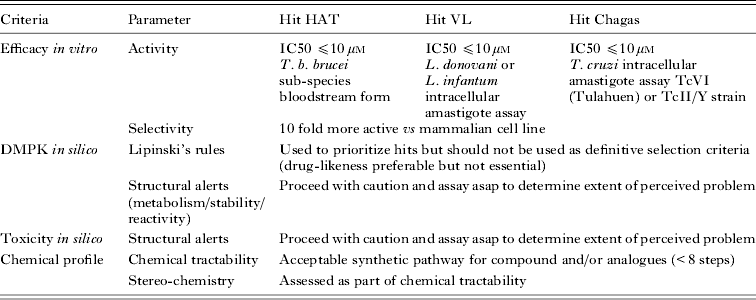
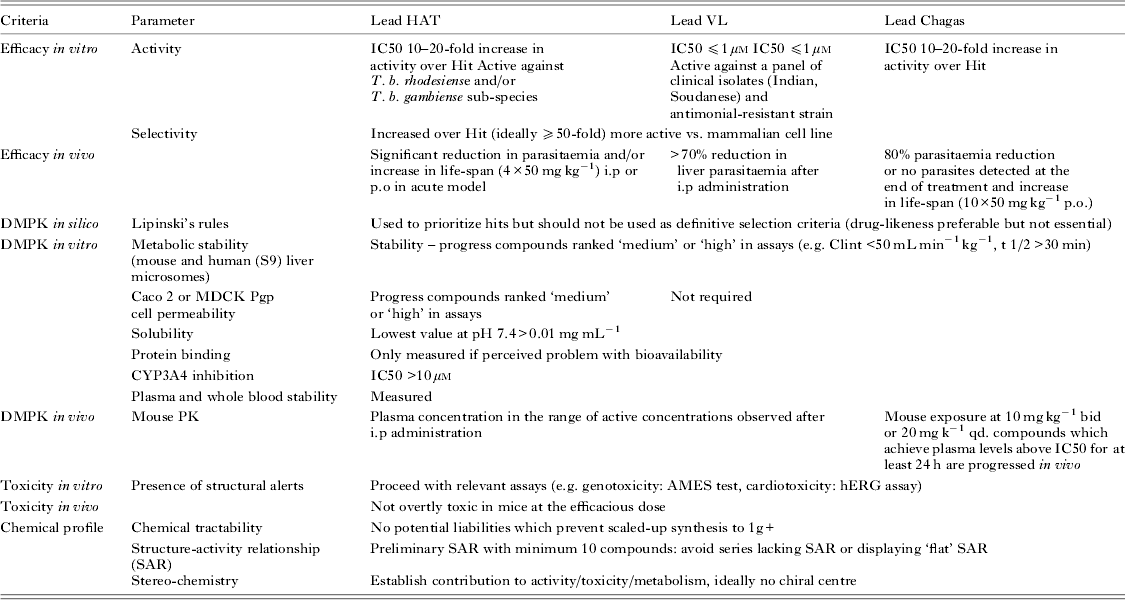
DNDi collaborates closely with its industrial partners, and has built new research partnerships with a number of pharmaceutical and biotechnology companies over the last 18 months in order to access chemical diversity (Ioset and Chang, Reference Ioset and Chang2011). Not only does this enable access to quality chemical libraries, but also, very importantly, to well annotated analogues after identification of hits from primary screens of small representative libraries. Most of our research collaborations have come with initial restrictions on publication of structural data from primary screens. Nonetheless, in line with DNDi's policy of open access, we are in positive negotiations with most partners from the pharmaceutical industry to make structures and annotation available as hits progress to lead optimization programmes, following the DNDi internally defined criteria and proposed cut-off values to qualify at the hit and lead selection stages (see Tables 1 and 2). It is worth noting that these are not applied strictly but are used to guide and prioritize hit and lead series, with the global activities, toxicities and chemical profiles being taken into account. The criteria and values listed are reviewed on a regular basis and are therefore subject to change.
Table 1. Criteria at hit stage

Table 2. Criteria at lead stage
Having gained access to considerable chemical diversity from the pharmaceutical industry, it has been necessary to carefully select or request specific libraries. Complete files belonging to large pharmaceutical companies are in the order of several million compounds and, as such, are still beyond the capacity for screening against intracellular amastigotes. The main strategies adopted by DNDi for library selection are outlined below.
Indication/repositioning libraries are small files of a few hundred compounds which have progressed to preclinical or clinical development for a variety of indications, with some compounds already registered as drugs. Screening such libraries could be considered to be very much a lottery, where the chance of success is very low but a positive hit would be associated with considerable annotation associated with a drug discovery programme. Unfortunately, no promising hits have yet been identified from screens of indication sets sponsored by DNDi.
A similar high-risk but potentially high-return strategy has been the screening of libraries which address specific pharmacokinetic, absorption, distribution, metabolism and excretion (PK/ADME) criteria. These include compounds which are orally available or which cross the blood-brain barrier, essential for drugs intended to treat human African trypanosomiasis. It is expected that any active compounds would qualify for, or be close to qualifying for, in vivo efficacy testing, and may also be related to a well characterized family of compounds held by the partner from the pharmaceutical sector. A small number of hits have been identified in these screens. In all cases, the human targets against which these compounds were developed did not have an equivalent parasitic target, and it is probable that the mechanism of action in the parasite is different.
Chemical classes with known anti-infective properties are the third class of compounds we selected for screening. These have included macrolides, fluoroquinolones, oxazolidinones, hydrazones, ribosome inhibitors and anti-fungal compounds amongst many other proprietary anti-infective compounds. None have so far yielded hits which have progressed past early lead optimization efforts. This probably reflects the absence or genotypic divergence of the kinetoplastid target from that in the infectious agents against which the original pharmaceutical research was directed. This can be illustrated by the differences in phylogenetic lineage and functional homology of DNA topoisomerase II from eukaryotic kinetoplastids as compared to other eukaryotic cells or prokaryotic bacteria, the latter being the target of quinolone antibiotics (Das et al. Reference Das, Dasgupta, Sengupta and Majumder2004). It should be noted, however, that this collaborative screening approach has not been exhaustively completed with the relevant pharmaceutical and biotechnology companies.
DNDi has sponsored a small number of phenotypic screens with target-based libraries such as protease inhibitors and kinase inhibitors. In general they have been avoided because of a lack of a thorough validation of these targets in spite of the considerable efforts of drug discovery directed towards them during the last few decades (Renslo and McKerrow, Reference Renslo and McKerrow2006; McKerrow et al. Reference McKerrow, Rosenthal, Swenerton and Doyle2008; Chawla and Madhubala, Reference Chawla and Madhubala2010). In addition, it is also likely that compounds with specificity towards the human orthologues of these targets will display reduced activity against the parasite target. This potential issue could eventually be addressed in the lead optimization phase in close collaboration with a pharmaceutical company via a selection of pre-existing moderately active or inactive analogues against the human target, as well as through the selective synthesis of analogues using in silico modelling tools supported by a counter screen on a human kinase panel.
Finally, diversity libraries have been selected as a source of new leads, such as the Pfizer Global Diversity Research Set which consists of 150 000 compounds representative of the global Pfizer 3·5 million collection (Bell et al. Reference Bell, Bradley, Everett, Knight, Loesel, Mathias, McLoughlin, Mills, Sharp, Williams and Wood2013). Several additional core diversity sets have also been accessed and screened by DNDi as part of ongoing collaborations with pharmaceutical companies. DNDi expects to identify most of their new compound classes for development from these and similar libraries, although low hit rates, lack of R&D background knowledge and IP encumbrance may present obstacles along the way. The global hit rate of so-called unbiased diversity sets is lower than 0·1% (ranging from 0·02 to 0·09%) when screening against the intra-macrophagic form of L. donovani, taking an activity cut-off of 10 μ m in dose-response determinations, calculated as IC50 combined with a 10-fold selectivity index versus a mammalian cell line. This hit rate was obtained from screening approximately 500 000 compounds from a variety of libraries, and is significantly lower than that from screening campaigns against the bloodstream form of T. brucei (average 0·45%, ranging from 0·1–1·2%, approximately 1·1 million compounds assayed) as well as the intra-macrophagic form of T. cruzi (average 0·30%, ranging from 0·05–0·85%, approximately 500 000 compounds assayed). The relatively wide data distribution related to the assay hit rates can mostly be explained by the use of various assay protocols and screening laboratories to operate the screens as well as by the nature of the compound collections submitted to these assays as each library has been designed using a distinct set of criteria.
Such a low hit rate for intracellular L. donovani screening, compared to that of the extracellular blood stream form of T. brucei sp., is not surprising. For a compound to reach a target in an intracellular parasite it must first pass through two host cell membranes and often hostile environments for a drug-like compound, including a pH of 5·4 and a range of cell host lysosomal enzymes in the parasitophorous vacuole (Lang et al. Reference Lang, Hellio, Kaye and Antoine1994). Moreover, intracellular amastigotes of L. donovani are slow-replicating pathogens (De Rycker et al. Reference De Rycker, Hallyburton, Thomas, Campbell, Wyllie, Joshi, Cameron, Gilbert, Wyatt, Frearson, Fairlamb and Gray2013) compared to intracellular amastigotes of T. cruzi, and have developed very sophisticated strategies to survive using a reduced metabolism within the host cell defence system (Kima, Reference Kima2007; Pescher et al. Reference Pescher, Blisnick, Bastin and Spath2011). Also, by hiding in the vacuolar compartment it is difficult for drugs to reach the parasites and exert their effect on biochemical pathways or targets that are still functional and essential to the survival of the L. donovani amastigote pathogen.
Not all hit compounds which come out of the screening process continue through to lead optimization: a significant number are dropped following analysis and careful assessment of the identified molecules, due to chemical clustering, duplication with ongoing and past hit-to-lead and lead optimization programmes at DNDi or elsewhere, and incompatibility with the current internal target product profiles defined from those diseases (www.dndi.org/diseases-projects/diseases.html). When the mechanism of action is not known, nor any preliminary structure-activity relationships associated with potency, it is usually safer to rely on hits associated with a high level of potency (ideally at the submicromolar level) in a phenotypic assay. Hit-to-lead and lead optimization experience has taught us that improving potency in the complex environments found in L. donovani or T. cruzi intracellular assays is a rather challenging task. Instead, optimization of PK/ADME is required to improve in vivo efficacy and build PK/PD knowledge within a specific series. Despite these hurdles, phenotypic screening is still considered more appropriate than target-based screening, and the fastest way to increase chemical diversity in a field where most targets are not fully validated and few known anti-infective classes of compounds are potent. To date, DNDi has identified an estimated total of over 50 chemical lead series from its screening and post-screening operations, with some, such as nitroimidazoles and oxaboroles, active against multiple protozoans. Two drug candidates emerging from this preclinical pipeline (www.dndi.org/diseases-projects/portfolio.html) are currently being progressed in clinical trials for human African trypanosomiasis (fexinidazole and SCYX-7158) and visceral leishmaniasis (fexinidazole).
FUTURE PERSPECTIVES
A large diversity within screened chemical libraries is most likely to yield the greatest number of new starting points for drug discovery. Currently, the major hurdle to this strategy is the technical (coupled to a financial) limitation in the capacity to screen against whole cell organisms, especially with respect to intracellular protozoa. Innovative approaches to improve throughput would relieve this bottleneck, and are currently under investigation. A recent manuscript (De Rycker et al. Reference De Rycker, Hallyburton, Thomas, Campbell, Wyllie, Joshi, Cameron, Gilbert, Wyatt, Frearson, Fairlamb and Gray2013) assessed the use of free living L. donovani axenic amastigotes as a surrogate predictive for intracellular amastigote activity. This comparative study demonstrated that from a screen of 16 000 diverse compounds in both axenic and intra-macrophage assays, with reconfirmation of the axenic hits in an intracellular assay, the hits obtained from the intracellular screen were also identified in the axenic amastigote screen. The axenic pre-screen identified a large number of positives that did not translate to the intracellular screen (false positives) but reduced the size of the library for confirmatory screening by more than 95%. Thus, a pre-screen against the extracellular form of the L. donovani parasite, followed by reconfirmation of the primary actives in the intracellular amastigote form of the assay, could offer a valuable cost-effective screening cascade when dealing with very large pharmaceutical company libraries. Such an approach could reasonably be applied to several global files from pharmaceutical companies. Indeed, despite difficulties in estimating the overlap in the chemical diversity between collections from Big Pharma, a recent precompetitive analysis of AstraZeneca (2·5 million) and Bayer Pharma AG (1·4 million) compound collections based on 2D near neighbour molecular fingerprinting highlighted only a limited overlap between the 2 compound sets (Kogej et al. Reference Kogej, Blomberg, Greasley, Mundt, Vainio, Schamberger, Schmidt and Hüser2013).
Screening larger diversity sets in association with an increasing capacity might not be the only way to secure new series for further development, however. At DNDi we intend to investigate, validate and eventually implement several alternative approaches to capture additional active scaffolds through an improved and more sophisticated selection of libraries. One attractive research avenue that we are currently exploring to significantly enhance the hit rate of screening programmes is the development of activity-predictive models, using public and proprietary data sets (the latter assumes partner's approval to use its IP in this framework has been secured). Such models have already been recently applied in the field of anti-infective discovery, notably in an Open Source context for tuberculosis and malaria (Periwal and Kishtapuram, Reference Periwal and Kishtapuram2012; Jamal and Periwal, Reference Jamal and Periwal2013). Although is it still too early to come to any definite conclusions regarding this innovative approach, predictive modelling is expected to assist the prioritization of compounds to be submitted for screening and drastically reduce the related screening expenses, whilst increasing the hit rate and identification of novel active series.
The development of classification techniques to compare and visualize collections of compounds, such as through a list of descriptors related or unrelated to the chemical structure of the compound sets using Principal Component Analysis (Feher and Schmidt, Reference Feher and Schmidt2003), without requesting the structural information to be shared among parties, could also be a valuable way to prioritize compound libraries for screening when collaborating with multiple pharmaceutical companies. Such tools could be used to share knowledge and identify potential opportunities to combine efforts at a later stage of drug development (e.g. across Product Development Partnerships preclinical programmes) without compromising confidentiality between the parties.
While obviously committed to honour any IP of its collaborating parties, DNDi also aims to share any derived knowledge with the scientific community active in the field of Neglected Diseases via its Open Innovation portal (www.dndi.org/diseases-projects/open-innovation.html). Just as GSK has previously published the 13 533 chemical libraries of compounds active against Plasmodium falciparum resulting from the screening of its 2 million compound library (Gamo et al. Reference Gamo, Sanz, Vidal, de Cozar, Alvarez, Lavandera, Vanderwall, Green, Kumar, Hasan, Brown, Peishoff, Cardon and Garcia-Bustos2010), DNDi has also deposited various data sets in the public domain, from screening and lead programmes. These include the screening of the Medicines for Malaria Venture (MMV) malaria box (www.mmv.org/malariabox), an exciting and innovative initiative to boost drug discovery in the field of neglected disease. More data will follow as publication authorization is granted by the collaborating parties.
Finally, the few tractable lead series which have been identified to date should be fully exploited by using them to conduct in silico screens against diversity libraries from different sources. DNDi is working with a number of pharmaceutical companies to develop strategies to conduct such screens against their libraries with each using their own proprietary screening algorithms and avoiding contamination of their intellectual property. This should maximize the diversity of scaffolds and annotation around a single series identified from a phenotypic screen.
ACKNOWLEDGEMENTS
The authors would like to thank Dr Sue Wells for the critical reading of the manuscript.
FINANCIAL SUPPORT
The Drugs for Neglected Diseases initiative (DNDi) is grateful to the following donors for funding its screening programmes from 2006 to date: Spanish Agency for International Development Cooperation (AECID), Spain; Department for International Development (DFID), United Kingdom; Ministry of Foreign and European Affairs (MAEE), France; Federal Ministry of Education and Research (BMBF) through KfW, Germany; GTZ on behalf of the Government of the Federal Republic of Germany; Swiss Agency for Development and Cooperation (SDC), Switzerland; Dutch Ministry of Foreign Affairs (DGIS), The Netherlands; Sasakawa peace Foundation; UBS Optimus Foundation; Sandoz Family Foundation; Medecins Sans Frontières (Doctors without Borders); Bill & Melinda Gates Foundation, United States of America. The donors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.