INTRODUCTION
Neurocysticercosis (NC), which is caused by the invasion of the central nervous system (CNS) with the Taenia solium metacestode (TsM), represents one of the principal public health problems worldwide and results in chronic morbidity and significant mortality (Bern et al. 1999; White, 2000). This disease primarily manifests with headache and seizure, but can also exhibit a highly protean set of clinical symptoms, depending on the number and location of the worms infecting the brain, in addition to the stage of the infection (Del Brutto, Sotelo and Roman, 1998; White, 2000). In endemic Latin American and African regions, NC accounts for 18–50% of adult-onset seizures in all admissions to neurological or psychiatric wards of local hospitals (Carpio, Escobar and Hauser, 1998; Palacio et al. 1998; Bern et al. 1999; Vilhena, Santos and Torgal, 1999; Phiri et al. 2003; Garcia et al. 2004). NC has also become an emerging or reemerging disease in industrialized countries, due largely to immigrants from and frequent travel to endemic areas (White, Robinson and Kuhn, 1997; Roman et al. 2000).
It is important to differentiate NC from other space-occupying lesions in the CNS, because the management plans instituted for such patients tend to be rather different and are normally tailored to idiosyncratic aetiology of each patient. In the case of NC alone, it is also important to determine the stage of the infection, as most cases in the early active stage can be successfully treated with specific chemotherapeutic agents, which have been demonstrated to ameliorate the clinical manifestations associated with local inflammation, as well as to reduce the incidence of late-onset refractory seizures in such patients (Baranwal et al. 1998; Del Brutto et al. 2001; Garcia et al. 2002, 2004). The detection of specific antibodies to the causative parasites has proven to be highly useful in confirming clinical diagnosis of NC, as have neuroimaging examinations. The identification of sensitive and specific antigens which allow species and stage differentiation is one of the daunting challenges faced by researchers in the scientific and medical communities.
The diagnosis of NC remains problematic, as the clinical presentation of the disease is frequently non-specific, and few standard descriptive symptoms and pathologies are available for guiding routine clinical examination (Del Brutto et al. 2001; Garcia et al. 2003). In addition, a host of difficulties remain to be solved, especially those associated with the lack of specificity of the currently available serological tests, as well as problems associated with standardization of their interpretations. Recent efforts have focused on identification of specific antigenic molecules and production of well-defined antigens with high purity and large quantities. The antigens that have been most extensively studied in TsM are a group of lentil-lectin purified glycoproteins (LLGP) with low-molecular weights ranging from 8–50 kDa, due to their relatively high sensitivity and specificity for serodiagnosis (Tsang, Brand and Boyer, 1989; Ito et al. 1998; Ko and Ng, 1998; Sako et al. 2000; Garcia et al. 2002; Villota et al. 2003).
In contrast, only few studies have been directed toward characterizing the macromolecular proteins in TsM. We previously described a highly antigenic and specific 150 kDa protein complex, which was composed of 3 subunits of 7, 10 and 15 kDa, in the TsM CF (Kim et al. 1986; Cho et al. 1988; Yang et al. 1998; Chung et al. 1999); however, it remains to be determined how many more macromolecular proteins exist in TsM other than the 150 kDa molecule, whether they consist of subunit proteins, and, if they are, the manner in which the subunits are linked, as well as which subunits are the most specific and useful in the context of serodiagnosis. The determination of these issues would clearly improve our ability to develop novel serodiagnostic assays.
The primary goal of the present study was to characterize the structural components of a 120 kDa glycoprotein complex in TsM CF by multiple proteomic approaches, including two-dimensional electrophoresis (2-DE), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), protein sequencing and recombinant protein expression. The proteomic analysis of TsM CF would prove invaluable, not only in understanding the relevant biochemical/biophysical properties and their physiological functions in the metabolism of the parasite, but also in further targeting of specific antigens. We also carried out a preliminary evaluation of the diagnostic value of the 120 kDa protein complex and its subunit components. In addition, we found evidence that the extensively studied TsM LLGP antigens (Tsang et al. 1989; Greene et al. 2000; Restrepo et al. 2000; Hancock et al. 2003) are associated, both structurally and immunologically, with this 120 kDa TsM protein complex.
MATERIALS AND METHODS
Collection of TsM CF
TsM was collected from naturally infected pigs in Palmar, Guyas Province, Ecuador. The CF was collected by puncturing the individual intact TsMs, followed by centrifugation at 20000 g at 4 °C for 1 h. The resulting supernatants were used as crude CF protein and were stored at −70 °C until use (Yang et al. 1998).
Serum and antibody samples
A total of 204 serum samples from NC patients were tested. The patients were diagnosed on the basis of their clinical manifestations, neuroimaging findings and elevation of specific antibody levels in their sera and/or cerebrospinal fluids (CSFs) as determined by enzyme-linked immunosorbent assay (ELISA) using crude TsM CF as the antigen (Cho et al. 1986). The patients were categorized into 3 groups according to their neuroimaging findings as previously described (Escobar, 1983). The patients whose scans indicated multiple low-density lesions (MLDs) or hydrocephalus consistent with NC were designated as the active cases (n=159). Those who exhibited MLDs with multiple calcifications (MCs) were designated as the mixed cases (n=25). Chronic inactive cases (n=20) were defined only when MCs were observed on their CT/MRI scans (Del Brutto et al. 1998; Chung et al. 1999). Serum samples from patients infected with adult T. solium (n=15), T. saginata (n=15) and T. asiatica (n=10) worms were also used. These patients were diagnosed on the basis of the morphological characteristics of the scolices and the gravid proglottids discharged, as well as the specific bands observed on random amplified polymorphic DNA (RAPD) analysis (Eom et al. 2002). In addition, the serum samples from patients with alveolar echinococcosis (AE, n=8), cystic echinococcosis (CE, n=50), sparganosis (n=30), paragonimiasis (n=30) and clonorchiasis (n=30) were included in this test. Normal healthy controls (n=50), all of whom denied any possible exposure to helminthic or protozoan infections were also tested.
Polyclonal rabbit antiserum against crude CF (RACF) was produced by the immunization of New Zealand white rabbits 3 times with CF, coupled with Freund's adjuvants. A monospecific antibody against the TsM 10 kDa protein and a monoclonal antibody against the TsM 150 kDa protein were obtained as was previously described (Kim et al. 1986; Chung et al. 1999). All serum and antibody samples used in this study were stored at −70 °C until use.
Fast performance liquid chromatography (FPLC)
CF proteins were fractionated by a Superdex 200 prep grade (HiLoad, 16×60 cm-long) molecular sieve FPLC system (AKTA, Amersham Biosciences, Piscataway, NJ, USA). The column was equilibrated with 20 mM Tris-HCl (pH 8·0) supplemented with 150 mM NaCl, after which 3 ml (10 mg) of crude CF were applied to the column, at a flow rate of 0·5 ml/min. A total of 85 fractions (each in 1·5 ml) were allocated according to their absorbance at 280 nm monitored by the computer program, UNICORN (ver3.0). Each of the fractions were analysed by native-polyacrylamide gel electrophoresis (PAGE), sodium dodecyl sulfate (SDS)-PAGE and immunoblotting. The column was pre-calibrated with gel-filtration molecular mass standards (Amersham Biosciences) to estimate the molecular mass of each protein peak.
Native-PAGE and SDS-PAGE
Native-PAGE was done as previously described (Blum, Beier and Grass, 1987). Samples were mixed with equal volumes of sample buffer (0·2 M Tris-HCl, pH 6·8, 10% glycerol, 0·02% bromophenol blue) and separated on 6% rod gels. SDS-PAGE was conducted with 15% gels under either reducing or non-reducing conditions. For the non-reducing SDS-PAGE, the samples were mixed with sample buffer (without β-mercaptoethanol) and loaded on the gel without boiling. Reducing SDS-PAGE was done in the presence of β-mercaptoethanol according to the standard method. The gels were stained with either Coomassie blue or silver nitrate, or were processed further by immunoblot analysis.
Immunoelectrophoresis (IEP)
IEP was performed on glass plates (Amersham Biosciences) covered with 0·9% agarose gel (Gibco-BRL, Grand Islands, New York, NY, USA) in a borate buffer (50 mM, pH 8·6). A total of 20 μl (150 μg) of crude CF and the partially purified protein (80 μg) were electrophoresed for 4 h at 20 mA in the first-dimensional agarose gel. The troughs were removed and filled with RACF at a 1[ratio ]100 dilution in phosphate-buffered saline (PBS; pH 7·4), followed by overnight incubation in a humidity-controlled chamber at 37 °C. The gel plates were extensively washed with PBS, pressed, dried and stained with Amido black B (Sigma, St Louis, MO, USA).
Two-dimensional electrophoresis (2-DE) and protein identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS)
Isoelectric focusing (IEF) was done using an IPGphor system (Görg et al. 2000). The purified 120 kDa protein (30 μg) was mixed with rehydration buffer containing 6 M urea, 2 M thiourea, 2% (w/v) CHAPS, 0·4% (w/v) DTT, 0·5% IPG buffer and 0·002% (w/v) bromophenol blue. The samples were loaded on IPG strips (pH 3–11 or pH 6–11) by in-gel rehydration and were focused for a total of 35 kVh. Second-dimensional electrophoresis was performed on 15% SDS-PAGE gels (160×160×1 mm) with a 40 mA constant current. The analytical gels were stained with silver nitrate, whereas the preparative gels were stained with colloidal Coomassie blue G-250.
Protein spots in the 2-DE gels were then excised and processed by in-gel trypsin digestion and the digests were subsequently prepared as targets for mass spectrometry via the solution-phase nitrocellulose method (Landry, Lombardo and Smith, 2000). The target spots were analysed by Voyager-DE STR MALDI-TOF mass spectrometry (PerSeptive Bio-systems, Framingham, MA, USA). The monoisotopic peptide masses were selected in a mass range of 900–2500 Da. A peptide map was generated via peptide mass fingerprinting (PMF) using the Mascot software (http://www.matrixscience.com), as well as the protein sequence database of the National Center for Biotechnology Information (NCBI; Bethesda, NJ, USA).
N-terminal amino acid sequencing
The proteins, after being resolved by 2-DE, were transferred to polyvinylidene difluoride (PVDF) membranes and stained with Coomassie blue. The spots were excised and then used for protein sequencing on an ABI model 477A protein sequencer and an ABI model 120A PTH-analyzer (Perkin Elmer Applied Biosystems, Branchburg, NJ, USA) at the Basic Science Institute (Daejeon, Korea).
Polymerase chain reaction (PCR), cloning and expression of recombinant protein
Two oligonucleotide primer pairs were designed, predicated on a multiple sequence alignment of the 14 and 18 kDa protein genes of TsM, which were retrieved from the GenBank database. The sense primers, all of which contained a Sal I site (underlined sequences) were 5′-GGAGTCGACGCAACAA ACCGAAAGATGTT-3′ (for 14 kDa) and 5′-GGCGTCGACACAACAAACCGAAGTGTGAT-3′ (for 18 kDa); the antisense primers with the Not I sites were 5′-CAGCGGCCGCCATTAAGCAGTTTTTTTCTT-3′ (for 14 kDa) and 5′-CAGCGGCCGCCATTAAGCAGTTTTGTTCTT-3′ (for 18 kDa). The coding regions of the 14 and 18 kDa protein genes were amplified separately from a TsM cDNA library (Chung et al. 1999) by PCR in a 50 μl reaction containing 10 mM Tris-HCl (pH 8·3), 50 mM KCl, 2·5 mM MgCl2, 0·2 mM dNTPs, 25 pM each primer, and 2·5 units of AmpliTaq DNA polymerase (Perkin Elmer, Foster City, CA, USA). PCR amplification was done in a DNA thermal cycler (M9600, Perkin Elmer) for 35 cycles with a denaturation step at 94 °C for 30 sec, annealing at 56 °C for 30 sec, and extension at 72 °C for 1 min, with a final extension at 72 °C for 5 min. The PCR products were then analysed by 1·5% agarose gel electrophoresis and were subcloned into pGEM T easy-vector (Promega). The nucleotide sequences were determined using the ABI Prism Dye Terminator Cycle Sequencing Core Kit (Perkin Elmer) and an automated DNA sequencer (Applied Biosystems model 373A; Perkin Elmer). Nucleotide and amino acid information were obtained with the DNA Strider (ver3.0), as well as the BLAST programs provided in the NCBI databases.
The inserts in the plasmids harbouring the expected coding sequences were excised by Sal I and Not I double digestion, then cloned into the pGEX-6P-1 expression vector (Amersham-Pharmacia Biotech, Uppsala, Sweden), which expresses the recombinant protein in fusion with glutathione S-transferase (GST). After verification of the sequences, the plasmid constructs were transformed into E. coli BL21 (DE3) cells. Upon induction with isopropyl-β-D-thiogalactoside, the cells were harvested by centrifugation and lysed by sonication. The supernatants were adsorbed to glutathione-Sepharose 4B resin (Amersham-Pharmacia) and the fusion proteins were eluted with 10 mM reduced glutathione.
Southern hybridization
The TsM genomic DNAs (10 μg each) digested using restriction enzymes including BamH I and Hind III were separated on 0·8% agarose gels and transferred to Hybond N+ membranes (Amersham Biosciences). The coding sequences of both the 14- and 18-kDa protein genes were amplified from each of the plasmid constructs by PCR using primers described above. During the reactions, amplicons were labelled with digoxigenin (Amersham Biosciences). The hybridization and the subsequent signal detection were performed using digoxigenin related reagents, under the conditions mandated by the manufacturer (Amersham Biosciences).
Molecular phylogenetic analysis
A search of the non-redundant database at the NCBI (http://www.ncbi.nlm.nih.gov) with the Blast N algorithm identified over 20 nucleotide sequences which encode for TsM 14 or 18 kDa related antigens. Of these, 12 sequences which exhibited significant degree of homology (>60%) were aligned with the CLUSTAL W program. After optimization and manual trimming of the alignment, sequence divergence and phylogenetic relationship were determined using DNADIST and NEIGHBOR, respectively, in PHYLIP package (ver3.5c). The phylogram was viewed with TreeView (Page, 1996). Bootstrapping analysis with 100 replicates was carried out to evaluate the statistical significance of each node.
Immunoblot and ELISA
The proteins were separated on 15% SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes (Schleicher and Schuell, Germany). The membranes were incubated overnight with either antibodies or serum samples. RACF was used at a 1[ratio ]5000 dilution and the patient sera were used at dilutions of 1[ratio ]200. Peroxidase-conjugated secondary antibodies, including anti-rabbit IgG and anti-human IgG (Cappel, West Chester, PA, USA), at a 1[ratio ]1000 dilution, were incubated with membranes for an additional 3–4 h. The reactions were developed using 4-chloro-1-naphthol as a chromogen (4C1N, Sigma).
For ELISA, 200 μl aliquots (2·5 μg/ml) were coated on microtitre plates (Costar Coning, Cambridge, MA, USA) in carbonate buffer (pH 9·6) overnight at 4 °C. Serum samples were diluted to 1[ratio ]100 in PBS containing 0·05% Tween 20 and incubated for 2 h at 37 °C, then with peroxidase conjugated anti-human IgG (heavy- and light-chain specific, Cappel) for another 2 h at 37 °C at a dilution of 1[ratio ]1000. The reaction was developed using 0·03% o-phenylene diamine as a chromogen. The specific antibody levels (absorbance) were measured at 490 nm using a microtitre reader (Bio-Rad M3550, Herculus, CA, USA). An absorbance value of 0·18, which was designated as the lower limit for a positive reaction and also as the upper limit of the means+2 S.D. of the normal controls, was considered as cut-off value for a positive result (Cho et al. 1986).
RESULTS
The FPLC fractionation of TsM CF revealed two major protein peaks, of which 120 kDa is a novel macromolecular protein
Crude TsM CF was fractionated by molecular sieve FPLC and 85 fractions were obtained. The elution profile displayed 4 main peaks, which were designated groups I to IV, between fractions 25 and 61. The 2 highest peaks, groups III and IV, were located between fractions 40 and 42 and fractions 48 and 50, respectively (Fig. 1A). SDS-PAGE analysis of the 85 collected factions revealed similar results to those obtained by the profiling (data not shown). Comparison of the elution profiles with the molecular weights of the standard proteins indicated that groups III and IV eluted proteins corresponding to sizes of 150 kDa and 120 kDa, respectively. Immunoblot analysis revealed that the group III protein reacted strongly with a monoclonal antibody against the TsM 150 kDa protein, as well as with a monospecific antibody against the TsM 10 kDa protein (data not shown). These results indicated that group III represents the previously described 150 kDa protein complex, which harboured the 7, 10 and 15 kDa proteins as its principal subunits (Kim et al. 1986; Cho et al. 1988; Chung et al. 1999).

Fig. 1. Single-step purification of a 120 kDa protein of TsM CF by Superdex 200 FPLC system. (A) Chromatogram obtained from 1 ml of CF. Groups I–IV represent each of the different fractions of proteins pooled according to their absorbance peaks at 280 nm. (B) Analysis of the eluted group IV protein by native-PAGE gel stained with Coomassie Blue, showing a single and strong band with high-molecular weight, as well as a faint band with low-molecular weight (marked by a closed circle). (C) Immunoelectrophoretic analysis of the group IV protein, indicating that the protein formed a single small arc against RACF, whereas the crude CF formed at least 3–4 major arcs. CF, crude cyst fluid; RACF, rabbit anti-serum generated against crude cyst fluid.
However, the 120 kDa protein enriched in group IV has not been characterized. Therefore, in this study, we focused on the characterization of both biophysical and immunological properties of this protein. When the group IV proteins (a pool of 48–50 fractions) were subjected to native-PAGE, we observed a single, strong band with a high-molecular weight, although we also noted a faint band with low-molecular weight (Fig. 1B). IEP analysis indicated that the protein formed a single inner immunoprecipitation arc against RACF, whereas the crude TsM CF was shown to form at least 3–4 arcs (Fig. 1C).
The TsM 120 kDa protein complex consisted of six subunits which were derived from the 14 and 18 kDa glycoprotein
The TsM 120 kDa protein was separated by non-reducing SDS-PAGE, resulting in 2 broad bands of 42–46 (component a) and 22–28 kDa (component b) as well as 1 minor band of 55 kDa. The separation of the components a and b under non-reducing conditions indicated that noncovalent linkage(s) exist(s) between these two components. Additional SDS-PAGE analysis under reducing conditions resulted in 7 bands, of 14, 16, 18, 22, 28, 38 and 66 kDa, respectively (Fig. 2A). The components a and b were separately isolated by electroelution and their subunit compositions were observed by reducing SDS-PAGE. The component a was resolved into 14, 16, 18, 22, 28 and 38 kDa, while component b was separated into 14, 16 and 18 kDa (lanes a and b of Fig. 2A, respectively), indicating that these subunits are linked by covalent bonds. The antigenicity of these subunits was examined by immunoblotting with a pooled serum of active NC (n=10). As shown in Fig. 2C, all of these subunits were found to be immunoreactive. All of these subunit proteins were stained with PAS reagent (Fig. 2C), suggesting the presence of a carbohydrate moiety. The 55 or 66 kDa proteins observed under non-reducing or reducing SDS-PAGE conditions (Fig. 2A) were not reactive with the pooled patient serum and were later confirmed by MALDI-TOF MS analysis to be a porcine albumin (Fig. 3A).

Fig. 2. Biochemical properties of the TsM CF 120 protein purified by FPLC. The proteins were resolved by SDS-PAGE under reducing (R) or non-reducing (NR) conditions, followed by analysis with either silver nitrate staining (A), immunoblot with RACF (B) or Periodic Acid Schiff's (PAS) staining (C). Panel A indicates that the 120 kDa protein was composed of 2 major broad bands of 42–46 (a) and 22–28 kDa (b), respectively. When these 2 bands were separately isolated by electroelution followed by reducing SDS-PAGE, the 42–46 kDa band was separated into 14, 16, 18, 22, and 38 kDa subunits (lane a), whereas the 22–28 kDa band was separated into 14, 16, and 18 kDa subunits (lane b). Panel B demonstrates the results of immunoblotting of the same proteins from panel A. All of these proteins, with the exception of the 66 kDa band, exhibited antibody responses against RACF as shown and the pooled serum of the 10 active NC cases (data not shown). Panel C shows the results of PAS staining of the purified 120 kDa protein. The protein, separated by 15% SDS-PAGE under reducing (R) or non-reducing (NR) conditions, was fixed in 7·5% acetic acid overnight at 25 °C and was incubated with 2·2% Periodic acid for 45 min at 4 °C. Glycosylation was visualized by Schiff's reagent for 1 h at 4 °C and destaining by 0·1 M HCl containing 1% (w/v) sodium metabisulfite at 25 °C.

Fig. 3. Identification of the subunit proteins of the TsM 120 kDa protein complex by 2-DE followed by MALDI-TOF MS. (A) The protein was initially separated by NL IPG strips at pH values from 6–11 and then by 15% reducing SDS-PAGE. (B) For MALDI-TOF MS analysis, monoisotopic peptide masses were selected between 900 and 2500 Da from PTM. Peptides generated by the trypsination of the protein spots were then separated by liquid chromatography and the resultant peptides were identified with the Mascot searching program and the NCBInr database.
To further characterize the structural components of the TsM 120 kDa protein complex, we performed 2-DE, MALDI-TOF MS and N-terminal sequencing. As the component proteins of the native TsM 120 kDa protein were determined to be located in the basic portion of the preliminary 2-DE experiment, we separated the proteins on non-linear IPG strips with pH values of 6–11 and observed that the subunit proteins migrated between the pH values of 8·8–9·7 (Fig. 3A). In order to more precisely delineate the subunit molecules, 12 protein spots were excised from the 2-D gel and analysed by MALDI-TOF MS. The peptides generated by the trypsinization of the spots were separated by liquid chromatography and analysed via tandem mass spectrophotometry. Fig. 3B shows an example of a peptide mass fingerprint. Spots 1, 2, 5, 10 and 11 were identified as homologues of the TsM 14 kDa antigen (GenBank Accession no. AF257776), whereas spots 7 and 8 were determined to be homologues of the 18 kDa antigen (AF350070) (Table 1). However, spots 3, 4, 6, 9 and 12 were unidentifiable, due primarily to their low sequence coverage, which was, in turn, probably attributable to the paucity of the TsM genomic database. It was also possible that the proteins responsible for these spots possess complicated structures, or are tightly associated with other molecules. The N-terminal amino acid sequences of the spots 1, 2, 5, 10 and 11 were identical and were determined to be EKNKP, which is completely consistent with that of the 14 kDa antigen.

Cloning and phylogenetic analysis of the genes encoding for the TsM 14 and 18 kDa proteins
Based on both the N-terminal amino acid sequences of the 120 kDa subunit proteins and the nucleotide sequences encoding for TsM low molecular weight proteins in the GenBank, we designed a set of primers to clone the genes encoding the 14 and 18 kDa proteins. The PCR amplification of the TsM cDNA library using these primers allowed us to isolate the cDNA sequences encoding the mature portion of the 14 and 18 kDa proteins. The sequences were deposited in the GenBank database under the Accession numbers AY937272 for the 14 kDa and AY937273 for the 18 kDa protein. To determine the phylogenetic relationships between the identified 14 and 18 kDa genes, nucleotide sequences exhibiting significant homology with these two genes were subjected to multiple alignments (Fig. 4A). We found that the sequences obtained for the 14 and 18 kDa proteins were most closely related to the cDNA sequences, AF257776 (74% identity) and AF044081 (78% identity), or the genomic sequences, AF356337 and AF356345, respectively. Each of the genomic sequences was found to harbour 3 exons, interpolated with 2 introns (Fig. 4A). These 2 genomic sequences shared a substantial degree of sequence identity (78%) at the nucleotide level.

Fig. 4. Genomic structures and phylogenetic analysis of 14 and 18 kDa subunit genes. (A) The nucleotide sequences of the 14 and 18 kDa protein genes cloned in this study (TsM-14 and TsM-18, respectively) revealed high levels of sequence homology with genomic sequences, AF356337 and AF356345, and with mRNA sequences of AF257776 and AB044081. These genes consisted of 3 exons (indicated by upper cases) and 2 introns (shown by lower cases). Nucleotide sequences encoding a hydrophobic signal peptide are boxed in the 5′-end. The translation initiation and stop codons are doubly underlined. The polyadenylation signal is indicated by single underline. The predicted mature proteins of the 14 and 18 kDa were composed of 66 and 67 amino acids, respectively. Different nucleotides between the 14 and 18 kDa protein genes are highlighted in grey boxes and the sequence homology between them is indicated at the end of the alignment. (B) Phylogenetic analysis demonstrated that the TsM 14 and 18 kDa protein genes clustered separately into 2 tightly conserved clades, each of which included several isoform/variant genes. The phylogram was constructed by neighbour-joining algorithm and rooted with the 10 kDa gene as an out-group. Bootstrap values of over 50 were presented at each branching node to estimate the statistical significance of each node.
When we assessed the genomic distribution of the gene by Southern blot analysis, we observed multiple bands in the genomic DNAs which had been digested with the several restriction enzymes used (data not shown). These observations suggested a possibility of a multigene family, although the exact distribution patterns of each gene could not be differentially determined, mainly because of the high level of conservation between the nucleotide sequences of the genes. The phylogenetic analysis of 12 sequences closely related to the TsM 14 and 18 kDa proteins revealed 2 tightly conserved clades, each of which contained several isoform/variant genes (Fig. 4B).
Antibody reactivity of the purified native TsM 120 kDa protein and its subunits
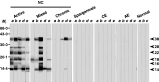
To evaluate the antibody responses, we employed immunoblot assays using FPLC-purified TsM native 120 kDa protein and sera obtained both from NC patients and other parasitic infections. The typical immunoblotting outcomes were shown in Fig. 5 and the antigenicity of each subunit protein was summarized in Table 2. Each protein manifested a distinct antibody recognition profile, with positive reaction rates ranging from 50 to 93%, and 30 to 80% against the sera from active and mixed stage NC, respectively. Among the 6 subunits, the 14 and 18 kDa proteins appeared to be the most sensitive and specific. We observed no cross-reactions with sera from patients with AE, CE or sparganosis, except for the 38 kDa protein, which was associated with cross-reactivity in 1 of 10 sera from the sparganosis patients.

Fig. 5. Immunoblot analysis of the FPLC-purified TsM 120 kDa protein complex against the serum samples from NC patients, other parasitic infections and from the normal controls. The proteins were separated by 15% reducing SDS-PAGE and transferred to nitrocellulose membranes. Each strip was incubated with an individual serum sample (a to e), as indicated on the top of each panel. CE and AE represent the cystic and alveolar echinococcoses, respectively. Protein molecular weight standards (Mr in kDa) are indicated at the left and the position of the 6 subunits of the 120 kDa proteins at the right (arrows).

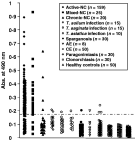
We further evaluated the serum antibody reactivity of the 120 kDa protein complex by ELISA employing a substantial panel of patient sera (Fig. 6). The sensitivity of the 120 kDa protein was found to be 85·5% and 72% against the sera obtained from the active and mixed NC patients, respectively. This protein exhibited a relatively low sensitivity (35%) to the sera from chronic NC patients. None of the sera acquired from the paragonimiasis or clonorchiasis patients, nor any of the sera from healthy controls showed positive antibody reactions. However, 3·7 to 12·5% of the sera from patients with AE, CE and sparganosis revealed cross-reactions. The overall sensitivity and specificity of the purified native TsM 120 kDa antigen were 84% and 97%, respectively (Table 3).

Fig. 6. Results of ELISA using the FPLC-purified TsM 120 kDa protein. The cut-off absorbance at 0·18 is indicated by the long horizontal dotted-line.

Evaluation of the diagnostic values of the recombinant TsM 14 and 18 kDa protein by immunoblot assay
We expressed the 14 and 18 kDa proteins (r14 and r18, respectively) as GST fusion forms in E. coli and purified them to homogeneity. These purified GST fusion proteins were directly used in immunoblot assay because we found that the GST domain did not affect the antibody reactivity of the recombinant proteins (data not shown). Representative results of the immunoblot assays are shown in Fig. 7. Positive antibody reactions against r14 and r18 were observed in 90% and 85% of the serum samples from the active and mixed stage NC (18/20 and 17/20 cases), whereas 2 and 1 of the 10 samples from chronic NC revealed positive reactions. In addition, 1 of each of the samples from the CE and sparganosis patients exhibited a weak cross-reaction against r14 and r18. None of the sera from other infections and normal healthy controls showed positive reactions. Overall specificity was determined to be 97·1% (34/35 cases).

Fig. 7. Immunoblot analysis of the antibody responses to r14 and r18 kDa protein in the serum samples from various helminthic infections, and those obtained from healthy normal controls. Two strips, each containing r14 and r18, were simultaneously incubated with the same serum samples, at 1[ratio ]100 dilutions. CE and AE represent cystic and alveolar echinococcosis, respectively.
DISCUSSION
In this study, we purified and characterized the 120 kDa protein complex in TsM CF. This complex is comprised of 6 subunits, which vary in size from 14 to 38 kDa. These molecules are linked principally by inter-/intra-molecular disulfide bonds and are modified by glycosylation to form the secondary and tertiary structures. These 6 subunits are found to be derived from either the 14 or 18 kDa backbone proteins. The detection of antibody reactivity to the purified 120 kDa antigen, to its subunits and to the recombinant forms of the 14 and 18 kDa subunits appears to be useful for serodiagnosis of both active and mixed-stage NC. Based on its primary sequence, biophysical structure and immunoreactivity profile, it appears to be likely that the subunits of the 120 kDa protein complex identified in this study represent some of the lentil-lectin purified glycoproteins (LLGP) with low-molecular weights, which have been described in many previous studies (Tsang et al. 1989; Greene et al. 2000; Restrepo et al. 2000; Sako et al. 2000; Hancock et al. 2003).
A panel of low molecular weight proteins in TsM have previously been identified and/or purified, using affinity columns with either monoclonal antibodies (Kim et al. 1986; Cho et al. 1988) or lectin Lens calinaris (Tsang et al. 1989; Restrepo et al. 2000; Haslam et al. 2003), or by preparative isoelectric focusing (Ito et al. 1998; Ko and Ng, 1998). However, the biochemical properties possessed by these antigens remain only incompletely understood, which limits their widespread use as antigens for routine serodiagnosis. It has also been reported that some of the LLGP, particularly those comprising the three major bands between 7 or 14 and 18 or 26 kDa, are regularly spaced and are frequently recognized simultaneously in the sera of NC patients (Restrepo et al. 2000; Hancock et al. 2003; Villota et al. 2003). These observations have raised questions of how many macromolecular proteins exist in TsM, what are their structural components and which components are useful for serodiagnosis. A few experiments have been conducted to resolve these issues. Two previous studies have shown that the chemical oxidation and enzymatic deglycosylation of the 12, 16 and 18 kDa TsM antigens resulted in a similarly sized protein backbone of approximately 7 kDa (Obregon-Henao et al. 2001; Haslam et al. 2003). This result suggests that the variable size of these three antigens is associated with different degrees of glycosylation. There is also a body of emerging evidence to suggest that a family of genes exists which encode for the LLGP in TsM (Greene, Wilkins and Tsang, 1999; Hancock et al. 2003). However, all of these approaches have focused on the characterization of the diagnostic value of each subunit protein, while they did not address the structural and serological relatedness of these antigens.
In the present study, we have described a method based on single-step FPLC, which allows efficient purification of the 120 kDa TsM antigen under native conditions. Our method is simple and effective, and generates a highly purified antigen in its native conformation, with an excellent recovery rate. This is quite different from most of the previously described purification processes, which were performed with complicated protocols (Tsang et al. 1989; Restrepo et al. 2000; Obregon-Henao et al. 2001) or under denaturing conditions (Ito et al. 1998; Ko and Ng, 1998). The availability of the highly purified 120 kDa TsM antigen in its native conformation allowed us to perform extensive proteomic analyses of this protein. Although a small amount of porcine serum albumin was determined to have contaminated the protein preparations, it was not associated with any interference in the structural, biochemical, or immunological protocols performed in this study.
We demonstrated that the 120 kDa TsM protein consisted of 2 primary components of 42–46 (component a) and 22–28 kDa (component b). These two components share 3 smaller subunits (14, 16 and 18 kDa), whereas the component a was found to harbour 3 additional subunits (22, 28 and 38 kDa). These 6 subunits appear to be linked by disulfide bonds and formed a macromolecular protein complex via covalent linking as analysed by reducing SDS-PAGE. The theorized disulfide linkage is further supported by the fact that both the 14 and 18 kDa protein sequences contain 1 and 2 cysteines, respectively. Two-DE and MALDI-TOF MS analyses revealed that the 14 and 18 kDa proteins may constitute the backbones of the 6 subunits of the 120 kDa protein. The 14, 22, 28 and 38 kDa subunits were found to be more closely related to the 14 kDa backbone, whereas the 16 and 18 kDa subunits appear to be more closely related to the 18 kDa backbone, according to analyses of their sequence similarities. Evidence for the presence of two different protein backbones is also supported by the observation that the sequences we cloned for the 14 and 18 kDa protein genes were closely related but were clearly different, with 82·3% nucleotide homology and 65·2% amino acid homology. The discrepancy between the apparent molecular mass determined by SDS-PAGE for these two proteins and that predicted from the deduced amino acid sequence (approximately 7 kDa for both), can be explained by differences in post-translational modification, including glycosylation and phosphorylation, as was demonstrated in previous studies (Restrepo et al. 2000; Obregon-Henao et al. 2001). Our preliminary PAS staining analysis indicates that all of the 6 subunits are glycosylated and this is further supported by the presence of one (positioned at the 10th Asn) and two (positioned at the 10th and 64th Asn) N-linked glycosylation sites in the 14 and 18 kDa protein sequences. The genes encoding for the 14 and 18 kDa proteins were also determined to harbour 2 (11th and 53rd Ser) and 3 (11th and 54th Ser and 12th Thr) putative phosphorylation sites in their primary sequences.
To date, more than 30 nucleotide sequences have been identified as genes which might encode for TsM LLGP. Previous phylogenetic analyses of 16 sequences encoding for TsM LLGP indicated 4 separate clades, designated Ts14, Ts18, TsRS1 and TsRS2 (Hancock et al. 2003). We attempted to characterize the phylogenetic relationship between the sequences we cloned for the 14 and 18 kDa proteins and determined that they belonged to the Ts14 and Ts18 clades. This finding further supports the notion of the existence of 2 distinct backbone proteins for the 120 kDa TsM protein complex. The results of Southern blot analyses indicate that both the 14 and 18 kDa proteins are encoded by multiple copy genes. Further studies are needed to identify additional sequences and to localize the genes on the chromosomes. The presence of variable sequences for both the 14 and 18 kDa proteins raises questions as to whether these two proteins, as identified by SDS-PAGE, represent a mixture of homozygous or of heterozygous molecules. Based on the results of 2-DE analyses, the 14 and 18 kDa protein bands each contained at least 8 or 9 spots, the pH values of which varied from 8·8 to 9·7, respectively. These spots may represent different members of the Ts14 and Ts18 clades. These data imply that the protein bands observed in regular SDS-PAGE consist of heterozygous molecules. More comprehensive investigation into the contribution of each molecule to antibody reactivity, as well as the regulation of their expressions may prove vital to the development of novel serological tests.
To determine the usefulness of the TsM 120 kDa antigen for the serodiagnosis of NC, we examined the antibody reactivity of the purified protein as a whole molecule, in addition to its individual subunits. When the purified 120 kDa antigen was used directly in ELISA, we observed that it exhibited similar sensitivity, but higher specificity compared to the crude TsM CF antigen. The lack of antibody reactivity with the sera from patients with adult T. solium infection would appear to indicate that the 120 kDa antigen exhibits stage specificity. To evaluate the contribution of each subunit protein to the immunoreactivity of the 120 kDa antigen, we separated these subunits by reducing SDS-PAGE and determined the extent of their antibody reactivity by immunoblotting. Among the 6 subunits, the 14 and 18 kDa proteins were found to be most sensitive in detecting active and mixed stage NC cases. The 28 kDa subunit showed the lowest. All subunits except the 14 and 38 kDa subunits did not exhibit antibody reactivity with the sera from patients with chronic NC. Cross-reactivity with the sera from patients infected with other parasites, including the closely related parasite, Echinococcus, was minimally observed. When any of the 6 subunits exhibited positive antibody reactivity, the overall sensitivity and specificity were found to be 94·0% and 96·7%. These values appear to be similar to those obtained by ELISA with the purified 120 kDa native protein complex, and were also compatible with those of the gold standard immunoblot assay for NC serodiagnosis (Tsang et al. 1989; Garcia et al. 2002).
As the 14 and 18 kDa subunits are the backbone proteins of the 120 kDa protein complex, we expressed both subunits in an antigenically active form and evaluated their diagnostic values by immunoblotting. Both the sensitivity and specificity of the recombinant proteins appear to be consistent with those of the immobilized native antigens in detecting active and mixed stage NC. We are currently testing the viability of using these two antigens, either individually or combined as an antigen cocktail, within a simple and quantitative ELISA format. We are also planning to do seroepidemiological studies using these recombinant antigens in endemic areas. More detailed understanding of the structural and immunological relatedness of these specific antigens will clearly facilitate the development of novel serodiagnostic assays for NC.
E.-G. Lee and Y.-A. Bae contributed equally to the work. This work was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (02-PJ1-PG3-20204-0006).