INTRODUCTION
Sarcocystis neurona is an apicomplexan protozoan terrestrial parasite with a complex life cycle that includes sexual reproduction in a definitive host and asexual multiplication in intermediate hosts. The North American opossum (Didelphis virginiana), a non-native California terrestrial marsupial, as well as the South American opossum species (Didelphis albiventris), serve as the only known definitive hosts for S. neurona (Fenger, Reference Fenger1997; Dubey et al. Reference Dubey, Lindsay, Kerber, Kasai, Pena, Gennari, Kwok, Shen and Rosenthal2001a ). Following sexual reproduction within the gastrointestinal tract of the opossums, sporocysts are released into the environment in feces (Fenger et al. Reference Fenger, Granstrom, Langemeier, Stamper, Donahue, Patterson, Gajadhar, Marteniuk, Zhou and Dubey1995; Dubey et al. Reference Dubey, Lindsay, Saville, Reed, Granstrom and Speer2001b ). Susceptible intermediate hosts can become infected through oral ingestion of sporocysts, after which systemic parasite proliferation can induce inflammation in muscle and brain (Dubey et al. Reference Dubey, Rosypal, Rosenthal, Thomas, Lindsay, Stanek, Reed and Saville2001c ; Thomas et al. Reference Thomas, Dubey, Lindsay, Cole and Meteyer2007; Rejmanek et al. Reference Rejmanek, Miller, Grigg, Crosbie and Conrad2010).
Sarcocystis neruona was first discovered to cause a fatal disease known as equine protozoal myeloencephalitis (EPM) in horses in the USA (Dubey et al. Reference Dubey, Davis, Speer, Bowman, Delahunta, Granstrom, Topper, Hamir, Cummings and Suter1991). Although only terrestrial hosts (opossums) are known to serve as definitive hosts for S. neurona, this parasite has been implicated as an important cause of protozoal encephalitis in threatened southern sea otters (Enhydra lutris nereis) (Thomas et al. Reference Miller, Conrad, Harris, Hatfield, Langlois, Jessup, Magargal, Packham, Toy-Choutka, Melli, Murray, Gulland and Grigg2007; Miller et al. 2010). In addition to causing mortalities in sea otters, S. neurona has also been isolated and documented to cause systemic infection as well as fatal encephalitis in other marine mammal species [as reviewed by Dubey et al. (Reference Dubey, Howe, Furr, Saville, Marsh, Reed and Grigg2015)]. Published reports in marine mammals include S. neurona infections in Pacific harbour seals (Phoca vitulina richardsi), harbour porpoises (Phocoena phocoena), California sea lions (Zalophus californianus), northern fur seals (Callorhinus ursinus), Guadalupe fur seals (Arctocephalus townsendi), northern elephant seals (Mirounga angustirostris), Steller sea lions (Eumatopias jubatus), pygmy sperm whale (Kogia breviceps), killer whale (Orcinus orca) and Pacific white-sided dolphin (Langenorhynchus obliquidens) (Lapointe et al. Reference Lapointe, Duignan, Marsh, Gulland, Barr, Naydan, King, Farman, Huntingdon and Lowenstine1998; Miller et al. Reference Miller, Sverlow, Crosbie, Barr, Lowenstine, Gulland, Packham and Conrad2001; Rejmanek et al. Reference Rejmanek, Miller, Grigg, Crosbie and Conrad2010; Gibson et al. Reference Gibson, Raverty, Lambourn, Huggins, Magargal and Grigg2011; Carlson-Bremer et al. Reference Carlson-Bremer, Gulland, Johnson, Colegrove and Van Bonn2012; Barbosa et al. Reference Barbosa, Johnson, Lambourn, Gibson, Haman, Huggins, Sweeny, Sundar, Raverty and Grigg2015).
Despite the detrimental impact of S. neurona on the health of the threatened southern sea otter population, an understanding of factors influencing S. neurona transport to the nearshore marine environment, as well as exposure routes to otters, remain unknown. Epidemiological investigations have suggested that freshwater discharges occurring during the rainy seasons could deliver S. neurona sporocysts to marine waters, as sea otter deaths due to this parasite have been reported to occur predominantly in Spring months and near freshwater outflows (Kreuder et al. Reference Kreuder, Miller, Jessup, Lowenstein, Harris, Ames, Carpenter, Conrad and Mazet2003; Miller et al. Reference Miller, Conrad, Harris, Hatfield, Langlois, Jessup, Magargal, Packham, Toy-Choutka, Melli, Murray, Gulland and Grigg2010; Shapiro et al. Reference Shapiro, Miller and Mazet2012). A dietary link has also been proposed as a likely route of sea otter infection with terrestrial pathogens (Johnson et al. Reference Johnson, Tinker, Estes, Conrad, Staedler, Miller, Jessup and Mazet2009). To date, detection of S. neurona in either marine waters or shellfish has not been described. This study was designed to validate molecular assays for detection of S. neurona in mussels, and then apply the optimized polymerase chain reaction (PCR) methods in a surveillance field investigation to test for S. neurona contamination in wild mussels collected from central California. The hypothesis of the study was that mussel contamination with S. neurona would be highest in mussels that are: (i) sampled near freshwater runoff, and (ii) collected during wet season months.
MATERIALS AND METHODS
Mussel haemolymph spiking experiments
Spiking experiments were performed to determine the sensitivity of PCR assays for detection of S. neurona in the haemolymph of California mussels (Mytilus californianus). Thirty wild California mussels were obtained from intertidal rocks during low tide near Cambria, CA (35·5686, −121·1100), and transported on ice to UC Davis within 24 h. The shell of each mussel was cleaned using sterile brushes and water to remove any residual seawater and debris. Haemolymph fluid was aspirated from mussels by filing a notch and removing the fluid from the adductor muscle using a sterile 22-gauge needle. The haemolymph from all 30 mussels was combined to create a uniform matrix, and then reallocated into 1 mL aliquots for the spiking study.
Sarcocystis neurona sporocysts used in mussel haemolymph spiking experiments were previously obtained from opossum intestinal scrapings and stored in 1·5 mL microcentrifuge tubes at −80 °C (Rejmanek et al. Reference Rejmanek, Vanwormer, Miller, Mazet, Nichelason, Melli, Packham, Jessup and Conrad2009). Frozen intestinal pellets were thawed at room temperature, and then transferred to 15 mL conical tubes containing 10 mL bleach (final sodium-hypochlorite concentration 3%). Tubes were vortexed for 10 s, and placed on a rotating shaker for 15 min. Bleach-treated slurries where then subjected to two cycles of centrifugation, first using a fresh aliquot of 3% sodium-hypochlorite (5 min at 1000 × g , Sorvall RT7, Thermo Fisher Scientific, Waltham, MA), followed by a wash in phosphate-buffered saline (PBS) (10 min at 1000 × g ). The supernatant was removed and the pellet was transferred to a 1·5 mL microcentrifuge tube and subjected to two additional washes with PBS (5 min at 14 000 rpm, Eppendorf 5417C, Hauppauge, NY). The final pellet was resuspended in 1 mL PBS and an aliquot obtained for enumerating sporocysts using a haemocytometer chamber visualized under light microscopy. Microscopy also confirmed that prepared samples contained intact sporocysts with brightly fluorescing sporocyst walls under ultraviolet (UV) epifluorescence excitation, no fractured sporocysts or free sporozoites were observed. Serial dilutions of sporocysts were prepared and added to 1 mL aliquots of haemolymph in triplicate to achieve final concentrations ranging from 5 to 10 000 sporocysts/mL. Roughly 100 µL of PBS were added to three samples of haemolymph as negative controls.
Spiked haemolymph samples were then centrifuged (3 min at 14 000 rpm) and the pellet (100 µL) retained for nucleic acid extraction following previously published methods (Shapiro et al. Reference Shapiro, Mazet, Schriewer, Wuertz, Fritz, Miller, Largier and Conrad2010). In brief, 180 µL of ATL buffer (Qiagen, Valencia, CA) was added and the tubes were subjected to one cycle of freeze–thaw to release sporozoites from sporocyst walls by submersion in liquid nitrogen (5 min), followed by immediate transfer into a boiling water bath (5 min). Approximately 40 µL of proteinase K were added to each tube, followed by overnight incubation at 56 °C. Standard procedure for nucleic acid extraction was then followed using manufacturer instructions (Qiagen DNA mini kits®, Chatsworth, CA). Extracted DNA was amplified using the selected primer sets as described in Section 2.2.
Optimizing molecular assays for S. neurona detection in mussels
Amplification of S. neurona DNA in haemolymph was tested using five nested primer sets in a conventional (end-point) PCR assay (Table 1): the pan-apicomplexan gene, ITS-1 (Wendte et al. Reference Wendte, Miller, Nandra, Peat, Crosbie, Conrad and Grigg2010), ITS-1500 (~500 nucleotide sequence within the ITS-1 locus) (Miller et al. Reference Miller, Barr, Nordhausen, James, Magargal, Murray, Conrad, Toy-Choutka, Jessup and Grigg2009), snSAG3, snSAG4 (S. neurona-specific surface antigens) (Rejmanek et al. Reference Rejmanek, Miller, Grigg, Crosbie and Conrad2010; Wendte et al. Reference Wendte, Miller, Nandra, Peat, Crosbie, Conrad and Grigg2010) and the microsatellite gene sn7 (Asmundsson & Rosenthal, Reference Asmundsson and Rosenthal2006 #7). The PCR reaction mix (50 µL) used for all assays were modified from prior published protocols (Miller et al. Reference Miller, Grigg, Kreuder, James, Melli, Crosbie, Jessup, Boothroyd, Brownstein and Conrad2004; Rejmanek et al. Reference Rejmanek, Miller, Grigg, Crosbie and Conrad2010) and consisted of 36·1 µL (external reaction) or 39·1 µL (internal reaction) PCR grade water, 5 µL 10× PCR buffer with MgCl2 (15 mm), 1·0 µL dNTP mix (10 mm), 25 pm each of forward and reverse external primers (Table 1), 0·5 µL BSA (10%), 0·3 µL Taq polymerase (5 U/1 µL) and 5 or 2 µL template DNA in the external or internal reactions, respectively. A negative control consisting of PCR reagents and added water was included in all trials. The lowest limits of parasite detection for these markers were evaluated by assessing the lowest concentration of inoculated sporocysts that yielded detectable DNA amplification. DNA amplification conditions are summarized in Table 1.
Table 1. Thermocycler conditions and nested primer set sequences used in PCR assays for detection of Sarcocystis neurona in mussel haemolymph

Screening (ITS-1 and ITS-1500) assays and genotyping assays (snSAG3, snSAG4 and sn7) were validated through haemolymph spiking experiments using S. neurona sporocysts.
a All primer sequences are listed in 5′ to 3′ orientation. All PCR reactions were carried out in 50 µL reaction volumes with 5 µL of DNA template used in external reactions and 2μL DNA amplicon from the external reaction used in internal reactions.
Screening wild mussels for S. neurona
California mussels were collected from two field locations along the central California coast, near the towns of Carmel and Cambria, as previously described by Shapiro et al. (Reference Shapiro, VanWormer, Aguilar and Conrad2015). These locations were chosen based on prior investigations that identified them as high-risk areas for S. neurona-associated infections and/or deaths in sea otters (Miller et al. Reference Miller, Gardner, Kreuder, Paradies, Worcester, Jessup, Dodd, Harris, Ames, Packham and Conrad 2002 ; Shapiro et al. Reference Shapiro, Miller and Mazet2012). To further assess whether or not a temporal or spatial association existed for S. neurona contamination in bivalves, mussels were obtained during the wet and dry season (2011–2013) from two sites at each location, one proximal to freshwater runoff and one distant from (at least 5k) freshwater sources. The site proximal to freshwater at Cambria was Santa Rosa Creek (35·5686, −121·1100), and the site distant from freshwater was White Rock (35·5329, −121·0884). The site proximal to freshwater at the Carmel location was Carmel River Beach (36·5368, −121·9270), and the site distant from freshwater was Point Lobos (36·5166, −121·9583). At each sampling effort, 30 mussels were collected from each site and transported on ice to the laboratory. Within 24 h of collection, mussels were cleaned and haemolymph removed as described above. Haemolymph was centrifuged and the pellet (100–200 µL) subjected to nucleic acid extraction and DNA amplification as described above. Screening of mussel haemolymph for S. neurona was performed with the primer sets determined most sensitive for parasite detection using data from the spiking experiments: ITS-1 and ITS-1500. Further molecular characterization was attempted using the sn7 microsatellite primer set on all mussel samples confirmed to be positive for S. neurona.
In all PCR assays, tissue culture-derived S. neurona DNA was used as a positive control, and reagent blanks from nucleic acid extraction and PCR amplification procedures were used as negative controls. Amplified DNA products were run on gels (2% agarose) using electrophoresis and visualized using ethidium bromide under UV illumination. Samples that yielded DNA products consistent in size with S. neurona were further purified (Qiagen QIA quick Gel Extraction Kit) and submitted for nucleic acid sequence analysis (Division of Biological Sciences, DNA sequencing facility, University of California, Davis, CA). For sequence analysis, the forward and reverse DNA sequences were aligned using a multiple sequence comparison by log-expectation (MUSCLE) method via Geneious software (Biomatters, Auckland, New Zealand), ends trimmed, and the consensus sequence compared with GenBank reference sequences for S. neurona using the Basic Local Alignment Search Tool, BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
RESULTS
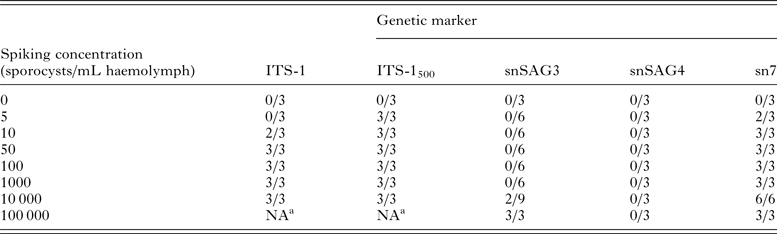
Spiking experiments demonstrated that the ITS-1500 primer set was the most sensitive assay for detection of S. neurona sporocysts in mussel haemolymph, with successful amplification of parasite DNA in all replicates containing ⩾5 sporocysts (Table 2). The ITS-1 and sn7 microsatellite primer sets yielded positive DNA amplification in two of three replicates containing 10 sporocysts and all replicates with ⩾50 sporocysts. PCR assays targeting single-copy gene sequences (snSAG3 and snSAG4) had low sensitivity for detection of S. neurona in mussel haemolymph (Table 2), with the snSAG3 primer set successfully amplifying the parasite only at a concentration of ⩾10 000 sporocysts per mL haemolymph. The snSAG4 primer set did not yield DNA amplification from any of the tested concentrations (up to 100 000 sporocysts).
Table 2. Number of positive replicate mussel haemolymph (1 mL) samples that tested positive for Sarcocystis neurona in validation spiking experiments using different concentrations of sporocysts
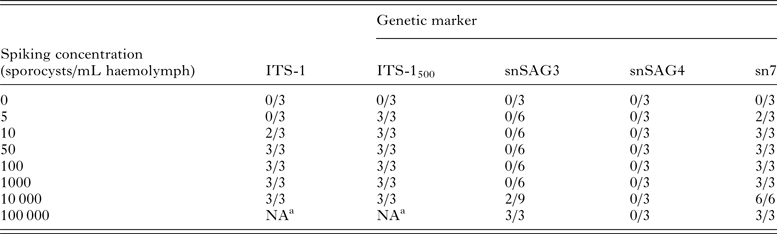
The multi-copy gene targets ITS-1 and ITS-1500 were evaluated for their use as screening assays, while snSAG3, snSAG4, and sn7 primer sets were tested to identify genotyping assays that could be used for further molecular characterization of S. neurona in mussels collected from the field.
a NA, not assessed.
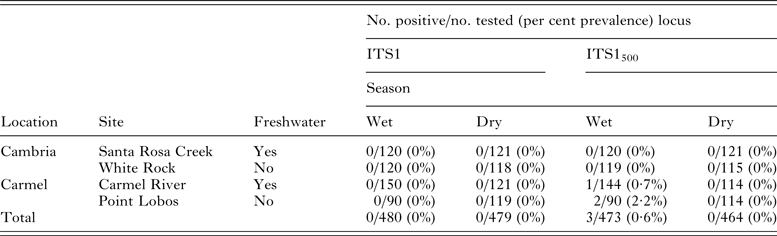
Following molecular assay validation through spiking experiments, a total of 959 and 937 wild mussels were screened for S. neurona DNA using the ITS-1 and ITS-1500 primer sets, respectively. Fewer samples were subjected to testing via the ITS-1500 primer sets due to insufficient DNA that could be used for both assays. No amplification products consistent with S. neurona were obtained using the ITS-1 primer set. In contrast, testing with the ITS-1500 assay revealed three mussels with amplified DNA consistent with S. neurona, which was subsequently confirmed via sequence analysis (Table 3). Of the three positive mussels, two were collected from Point Lobos (a site distant from freshwater) and one from Carmel River (a site proximal to freshwater), both within the Carmel field location. Neither sampling sites in Cambria yielded mussels with confirmed S. neurona DNA. All three S. neurona-positive mussels were collected during the wet season (April, 2013).
Table 3. Detection of Sarcocystis neurona DNA in wild-caught mussels collected from two field locations along the central California coast
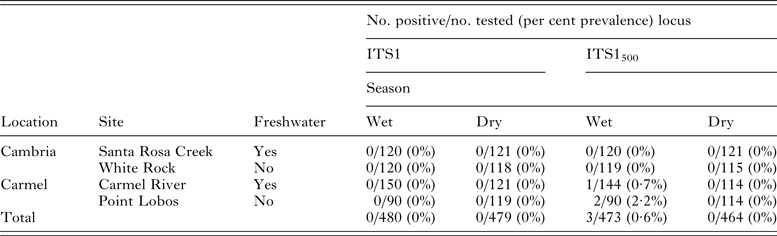
The three mussels that tested positive for S. neurona were all sampled in the Carmel region during the wet season (April 2013), with one mussel testing positive from Carmel River beach, and two mussels testing positive from Point Lobos.
DISCUSSION
This study is the first to validate sensitive molecular assays for the detection of S. neurona in mussels, and the first to report the presence of S. neurona in wild-caught mussels. Our findings support prior investigations that describe the ability of bivalves to filter and concentrate parasites from the environment (Miller et al. Reference Miller, Miller, Gardner, Atwill, Harris, Ames, Jessup, Melli, Paradies, Worcester, Olin, Barnes and Conrad2005; Shapiro et al. Reference Shapiro, VanWormer, Aguilar and Conrad2015 #2327), thus identifying mussels as sentinels for nearshore pathogen contamination. While the low number of positive mussels confirmed to be contaminated with S. neurona (3 of 959 tested) precluded statistical analysis, all contaminated mussels were sampled during the wet season following a rainfall event in April 2013, further supporting the hypothesis that rain-induced terrestrial runoff contributes to pathogen pollution in coastal ecosystems.
To maximize specificity and sensitivity of the molecular assays for detection of S. neurona DNA in mussels, the study evaluated five nested primer sets: ITS-1, ITS-1500, SnSAG3, SnSAG4 and one microsatellite marker, sn7. Haemolymph spiking trials determined that two screening assays, ITS-1 and ITS-1500, were sufficiently sensitive at detecting S. neurona DNA for application on field samples of wild-caught mussels. The ITS-1 primer set was chosen as a screening assay given its highly conserved nature and its ability to differentiate S. neurona from other Sarcocystis species, including Sarcocystis falcatula, which is also shed in the feces of opossums (Marsh et al. Reference Marsh, Barr, Tell, Bowman, Conrad, Ketcherside and Green1999; Tanhauser et al. Reference Tanhauser, Yowell, Cutler, Greiner, MacKay and Dame1999). Because of its highly conserved sequence, applying the particular ITS-1 primer set used in this study also provided an advantage of being able to screen for other parasites that can contaminate the nearshore marine environment, including Toxoplasma gondii, a related apicomplexan that also infects and kills threatened southern sea otters (Shapiro et al. Reference Shapiro, VanWormer, Aguilar and Conrad2015). A recent report by Shapiro et al. (Reference Shapiro, VanWormer, Aguilar and Conrad2015) describes the identification of T. gondii DNA in the same cohort of sampled mussels using the ITS-1 primer set. The spiking trials also revealed that the ITS-1500 primer set, that was originally developed by to detect and distinguish S. neurona from other Sarcocystis spp. in sea otter brain tissue (Miller et al. Reference Miller, Barr, Nordhausen, James, Magargal, Murray, Conrad, Toy-Choutka, Jessup and Grigg2009), was the most sensitive assay tested, consistently amplifying DNA in samples with ⩾5 sporocysts per mL haemolymph. Thus, this assay was applied as a second screening test for detection of S. neurona in wild caught mussels.
While the ITS-1 and ITS-1500 assays offer high sensitivity for parasite detection as they target a repetitive gene element, the highly conserved nature of this gene implies that it cannot be used to genetically discriminate among parasite isolates. Thus, further assays were evaluated as genotyping tools including a nested microsatellite primer set (sn7) and surface antigen (snSAG) genes. Prior molecular analysis using PCR confirmed a variety of polymorphisms among S. neurona genotypes using primers targeting microsatellite regions of the parasite's genome (Asmundsson & Rosenthal, Reference Asmundsson and Rosenthal2006; Rejmanek et al. Reference Rejmanek, Miller, Grigg, Crosbie and Conrad2010; Wendte et al. Reference Wendte, Miller, Nandra, Peat, Crosbie, Conrad and Grigg2010; Barbosa et al. Reference Barbosa, Johnson, Lambourn, Gibson, Haman, Huggins, Sweeny, Sundar, Raverty and Grigg2015). As the snSAG primer sets proved insensitive during spiking assays, only the Sn7 assay was applied to those mussels that tested positive with ITS-1500. However, DNA amplification was not successful in any of those samples.
The poor performance of the snSAG genotyping assays (detection limit of 10 000 sporocysts per mL haemolymph for snSAG3, and complete lack of parasite amplification for snSAG4 ⩽100 000 sporocysts) suggest that applying these assays for genotyping S. neurona on haemolymph from environmentally contaminated mussels is unlikely to be successful. One explanation for decreased sensitivity of these assays may be the presence of PCR inhibitors in mussel haemolymph. PCR inhibitors have previously been recorded in water samples and oysters used for molecular detection of T. gondii and viruses (Renault et al. Reference Renault, Le Deuff, Lipart and Delsert2000; Villena et al. Reference Villena, Aubert, Gomis, Ferte, Inglard, Denis-Bisiaux, Dondon, Pisano, Ortis and Pinon2004). While those studies showed that using BSA in the PCR assays improved recovery of T. gondii, inhibition of DNA amplification could still occur. Another reason that the surface antigen-targeting assays proved less sensitive may be due to these sequences only occurring as single copy genes, unlike the ITS-1 and microsatellite region which repeat within the genome of S. neurona, providing more DNA template to which the primers can anneal. The recent report by Barbosa et al. (Reference Barbosa, Johnson, Lambourn, Gibson, Haman, Huggins, Sweeny, Sundar, Raverty and Grigg2015) documenting a novel S. neurona genotype (XIII) that is particularly pathogenic in marine mammals demonstrates the need for developing sensitive molecular tools that can be used to genotype the parasite from environmental matrices in coastal ecosystems.
In addition to testing different molecular assays, applying PCR on mussel organs other than haemolymph may also be useful for optimizing DNA amplification. To date, no data have been reported on different bivalve tissues in which S. neurona is most likely to be detected in contaminated mussels. Future experiments that test the ability of bivalves to bioconcentrate S. neurona in different tissues through exposure of live mussels in aquaria would provide important insight for determining an optimal sampling testing approach for successful detection of the parasite. In this study, haemolymph was targeted for parasite detection due to the ease and efficiency of extracting the samples from nearly 1000 mussels. In addition, haemolymph has been shown as a suitable target for detection of the related terrestrial protozoal pathogen, T. gondii (Staggs et al. Reference Staggs, Keely, Ware, Schable, See, Gregorio, Zou, Su, Dubey and Villegas 2015 ). However, further studies are warranted to establish whether testing other bivalve tissues might yield a higher likelihood of S. neurona detection. For example, a study evaluating T. gondii oocyst retention in mussel samples reported that parasite DNA was detected in digestive gland more often than in haemolymph or gill tissue (Arkush et al. Reference Arkush, Miller, Leutenegger, Gardner, Packham, Heckeroth, Tenter, Barr and Conrad2003).
Successful amplification of nucleic acid products consistent with S. neurona was achieved in three wild-caught mussels. All three mussels positive for S. neurona were collected in the spring (April 2013), following rainfall events. Because of the low numbers of S. neurona positive mussels, statistical analysis to determine putative risk factors for mussel contamination with this parasite, such as collection during the rainy season or proximity to freshwater runoff, was not possible. Increased likelihood of S. neurona contamination in mussels sampled during the spring months may be linked to adult opossum shedding rates of S. neurona, which were reported to be highest during the reproductive months of March–July (Rejmanek et al. Reference Rejmanek, Vanwormer, Miller, Mazet, Nichelason, Melli, Packham, Jessup and Conrad2009). These findings are also consistent with previous sea otter mortality reports that identified a significant increase in otter morbidity and mortality due to S. neurona during the spring and following rainfall (Kreuder et al. Reference Kreuder, Miller, Jessup, Lowenstein, Harris, Ames, Carpenter, Conrad and Mazet2003; Shapiro et al. Reference Shapiro, Miller and Mazet2012). Miller et al. (Reference Miller, Conrad, Harris, Hatfield, Langlois, Jessup, Magargal, Packham, Toy-Choutka, Melli, Murray, Gulland and Grigg2010) further described an outbreak event of S. neurona mortalities in sea otters from Morro Bay in April of 2004, a month that followed several unusually high precipitation events in central California.
The hypothesis of a land to sea transport mechanism for terrestrial pathogens entering the coastal environment has also been raised for a closely related apicomplexan protozoan parasite, T. gondii (Conrad et al. Reference Conrad, Miller, Kreuder, James, Mazet, Dabritz, Jessup, Gulland and Grigg2005). Like S. neurona, sexual reproduction of T. gondii only occurs in a single group of terrestrial definitive hosts (domestic and wild felids), resulting in fecal excretion of the robust environmental stage of the parasite (oocysts) (Dubey et al. Reference Dubey, Miller and Frenkel1970; Hutchison et al. Reference Hutchison, Dunachie, Siim and Works1970). Miller et al. (Reference Miller, Gardner, Kreuder, Paradies, Worcester, Jessup, Dodd, Harris, Ames, Packham and Conrad2002) observed that otters in close proximity to overland runoff were three times more likely to acquire T. gondii, suggesting that oocysts were transported to the sea through freshwater sources.
One limitation of the molecular methods developed in this study for detection of S. neurona in environmental samples is that they cannot discriminate between DNA extracted from viable or non-viable parasites. Little information is available about the survival of S. neurona sporocysts in the nearshore environment, and parasite viability is likely to be impacted by multiple factors under natural environmental conditions. Variability in water temperature and salinity, UV light exposure, and physicochemical degradation within invertebrate tissues, may all contribute to sporocyst viability that will determine whether or not a sea otter or other susceptible animal becomes infected upon ingestion of a contaminated invertebrate. The related apicomplexan parasite, T. gondii, was determined to retain its viability for at least 3 days following contamination of mussels managed in the laboratory setting (Arkush et al. Reference Arkush, Miller, Leutenegger, Gardner, Packham, Heckeroth, Tenter, Barr and Conrad2003). Dubey et al. (Reference Dubey, Saville, Sreekumar, Shen, Lindsay, Pena, Vianna, Gennari and Reed2002) evaluated S. neurona sporocyst resistance to several physical and chemical stressors, and reported that sporocysts inoculated into mice were non-viable when subjected to temperatures >55 °C for at least 15 min, or when treated with ammonium hydroxide (29·5%) for >1 h. However, sporocysts remained viable when treated with Chlorhexadine (2%), Betadine (1%), bleach (10, 20 or 100%), as well as many other chemicals. While information on S. neurona survival in the marine environment has not been previously reported, the findings that identical parasite genotypes have been characterized in sea otters and opossums, sampled from adjacent coastal habitats (Rejmanek et al. Reference Rejmanek, Miller, Grigg, Crosbie and Conrad2010) suggest that sporocysts are able to survive the chemical and physical insults that are present during their transport from land to sea.
Results from this study demonstrate that marine bivalves can serve as an important sentinel for nearshore contamination with S. neurona. The finding that all three mussels that tested positive for S. neurona DNA were sampled during the wet season also supports prior studies reporting increased otter mortality due to S. neurona, as well as higher prevalence of opossums shedding sporocysts, during Spring months. The ability of mussels to concentrate sporocysts is also consistent with observational studies that report an association between S. neurona infection in otters and individual diet preference for clams and other soft sediment and bivalves (Johnson et al. Reference Johnson, Tinker, Estes, Conrad, Staedler, Miller, Jessup and Mazet2009). Thus, mussels may serve as important sources of S. neurona infection for threatened southern sea otters. The validated molecular tools developed here for S. neurona detection in mussels, particularly the sensitive primer set targeting the ITS-1500 locus, will provide researchers with useful tools to assist in unravelling additional dietary and environmental mechanisms by which this terrestrial parasite is transmitted to marine wildlife. Further insight on the environmental transmission mechanisms of S. neurona will aid the implementation of conservation and management decisions for reducing the load of terrestrial pathogens entering marine waters.
ACKNOWLEDGEMENTS
We are grateful to the field teams that supported field collections and laboratory processing of mussels, and specifically acknowledge Aiko Adell, Don Canestro, Miles Daniels, Mark Kocina, Colin Krusor, Zach Randell, Matt Smith, Tim Tinker, Joe Tomoleoni, Jim Webb, and Ben Weitzman. Laboratory processing of the mussels was further assisted by Terra Berardi, Heather Fritz, Leopoldo Guerrero, Kaitlyn Hanley, Claudia Llerandi, Ann Melli, Anna Naranjo, Andrea Packham, and Woutrina Smith. Collection of mussels from the sites in Cambria was facilitated through the University of California Ken Norris Rancho Marino Reserve.
FINANCIAL SUPPORT
We thank the UC Davis School of Veterinary Medicine STAR research programme for scholarship funding in support of L. Michaels, and The National Science Foundation (NSF) Ecology of Infectious Disease Programme (grant no. OCE-1065990) for funding this project (to KS and PC).