


I. INTRODUCTION
Voglibose, 5,6-didexoy-5-{[2-hydroxy-1-(hydroxymethyl) ethyl]amino}-1-C-(hydroxymethyl)-D-epi-inositol (as shown in Figure 1), has been the research focus due to its wide range of therapeutic and pharmacological properties, including its excellent inhibitory activity against α-glucosidase, hyperglycemia, and various disorders caused by hyperglycemia (Byrn et al., Reference Byrn, Pfeiffer, Ganey, Hoiberg and Poochikian1995; Giordano et al., Reference Giordano, Rossi, Moyano, Gazzaniga, Massarotti, Bini, Capsoni, Peveri, Redenti, Carima, Dagli Alberi and Zanol2001). Voglibose has shown strong antiobesity and antidiabetic activity as it is a new potent glucosidase inhibitor and is a drug used for type 2 diabetes in Japan and China (Morris et al., Reference Morris, Schlam, Chao and Short2000; Kariuki et al., Reference Kariuki, Zin, Tremayne and Harris1996). Voglibose obtained from organic synthesis processes is similar to the structure of the carbohydrates found naturally (Chen et al., Reference Chen, Zheng and Shen2006). So far, single-crystal voglibose was not synthesized under current working conditions, but single-crystal 5,6-dideoxy-5-{[2-hydroxy-1-(hydroxymethyl)ethyl]-amino}-1-C-(methoxycyclohexylmethyl)-D -epi-inositol can be synthesized as one of its by-products (Zhang et al., Reference Zhang, Sun, Ishurd, Pan and Ding2004). Voglibose was usually obtained as a polycrystalline material in industry production. In this paper, the crystal structure of polycrystalline voglibose determined by ab initio X-ray powder diffraction (XRPD) methods is reported.
II. EXPERIMENTAL
The sample was obtained in the process of voglibose synthesized by the method reported by Ohtake and Ikegami (Reference Ohtake and Ikegami2000). Then, the obtained sample was crystallized and purified for XRPD analysis.
Diffraction data were recorded on a X’Pert PRO MPD diffractometer (40 kV, 40 mA, and Cu K α radiation with λ=1.5406 Å), equipped with a diffracted-beam graphite monochromator, an X’Celerator scintillation counter, and an automatic divergence slit. Carefully ground powders were filled into a glass capillary with 0.7 mm in diameter. Other experimental conditions were step scan mode, with 5<2θ<130°, Δ2θ=0.02°, and t=10 s/step.
III. STRUCTURE SOLUTION
The first 20 diffraction peaks in the experimental X-ray diffraction pattern were first accurately estimated and then indexed using DICVOL program. The tile compound was determined to have a primitive triclinic cell with lattice parameters a=6.1974(6) Å, b=6.9918(5) Å, c=7.3955(9) Å, α=70.8628(3)°, β=103.5312(4)°, γ=94.3867(5)°, and V=294.2(2) Å3, and the values of figure of merit are M(20)=40, F(20)=43. The values of 2θ obs, 2θ cal, I/I o, h k l, d obs, and d cal are listed in Table I. A survey of the Cambridge Structural Database (version 2009) showed no match of any compound with these lattice parameters (or one derived therefore by standard cell-reduction programs). We, therefore, decided to solve the structure by ab initio XRPD methods using the experimental data.
Systematic absences, density, and geometrical considerations indicated Z=1 and space group P-1, which are later confirmed by successful structure solution and refinement. Table II contains the relevant crystal data and data analysis parameters. The structural model employed in the final whole-pattern Rietveld refinement was determined by the simulated annealing technique implemented in DASH3.0 (David et al., Reference David, Shankland, van de Streek, Pidcock, Motherwell and Cole2006). Based on the indexing results, the diffraction-peak intensities were first extracted by the Pawley refinement with values of figure of merit: R wp=16.0, R exp=9.68, and χ 2=2.52. During structure solving, there were 11 degrees of freedom including three positional coordinates for the centre of mass of the molecule, three parameters describing orientation of the molecule within the unit cell, and five torsions angles for C13-N12-C4-C3, C14-C13-N12-C4, O16-C10-C6-C5, O17-C14-C13-N12, and O18-C15-C13-N12.
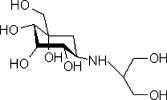
Figure 1. Structure formula for voglibose.
TABLE I. X-ray diffraction data for voglibose.
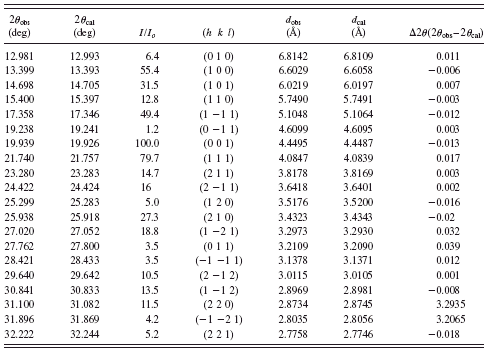
TABLE II. Crystal data and data analysis parameters for voglibose.

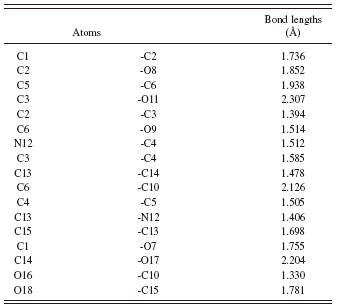
Simulating annealing finally ended with profile χ 2=18.80. The crystal and molecular structures are shown in Figure 2, and the final Rietveld refinement plots are plotted in Figure 3. As shown in Figure 2, the molecular structure of voglibose contains one six-membered ring, and the ring (from C1 to C6) adopts chair conformation. The 2-hydroxy-1-[(hydroxymethyl) ethyl] amino group aligned with C5 by the way of an axial bond. The angles for C6-C10-C5, N12-C4-C3, and C13-N12-C4 are 108.998°, 130.526°, and 158.723°, respectively. The final fractional atomic coordinates, bond angles, and bond lengths are listed Tables III – V, respectively.
IV. CONCLUSION
In the absence of single crystals, the ab initio structure of a polymorph voglibose was determined from conventional laboratory X-ray powder diffraction data. The crystal model

Figure 2. (Color online) Schematic illustration of voglibose molecule structure (left, ball and ticks; right, space filling model).

Figure 3. (Color online) Final Rietveld refinement plot (red line) with peak markers, raw experimental scan (blue line), and difference plot at the top.
TABLE III. Atomic coordinates for voglibose.

TABLE IV. Bond angles for voglibose.
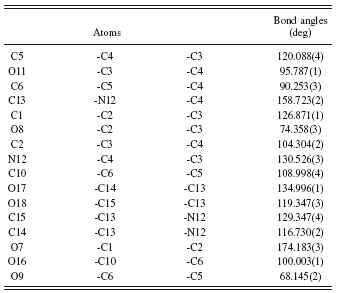
TABLE V. Bond lengths for voglibose.
was solved by simulated annealing using rigid groups with flexible conformational freedoms including five torsion angles, and the final crystal structure parameters, including atomic coordinates, bond angles, and bond lengths, were refined by rigid-body Rietveld refinement method.
ACKNOWLEDGMENTS
The authors would like to thank the financial supports from the Natural Science Foundation of Zhejiang Province (Grant No. Y4090167) and Key Laboratory of Advanced Textile Materials and Manufacturing Technology, Ministry of Education, Zhejiang Sci-Tech University.








