INTRODUCTION
The past several years has seen the global antimalarial pipeline bolstered with an influx of new chemical entities many acting through potentially unknown modes of action (http://www.mmv.org). This surge in new drug candidates can arguably be attributed to an increase in the number of high throughput screening (HTS) efforts by various academic and pharmaceutical research groups (Anthony et al. Reference Anthony, Burrows, Duparc, Moehrle and Wells2012; Guiguemde et al. Reference Guiguemde, Shelat, Garcia-Bustos, Diagana, Gamo and Guy2012). These phenotypic screening efforts have proven to be very successful as many of the compounds identified have been successfully optimized and progressed to preclinical development or beyond.
Historically antimalarial drug discovery efforts have focused primarily on targeting the asexual blood stages of the parasite; however, the next generation drugs not only need to represent new chemical classes with new modes of action (MOAs) to combat resistance but will also need to target sexual stages and the exo-erythrocytic stages (EEFs) of the parasite life cycle, or liver stages, as part of the eradication agenda. This becomes more important in the context of the growing threat and expansion of Plasmodium vivax malaria where primaquine remains the only approved treatment capable of clearing the dormant liver-stages of the parasite responsible for clinical relapse.
Drugs which act on one or more of the plasmodium life stages are known but rare. For example, atovaquone is active on blood and liver schizonts, while primaquine has activity on the dormant hypnozoites and the sexual stages or gametocytes (Delves et al. Reference Delves, Plouffe, Scheurer, Meister, Wittlin, Winzeler, Sinden and Leroy2012; Lelievre et al. Reference Lelievre, Almela, Lozano, Miguel, Franco, Leroy and Herreros2012). The recent development of in vitro assays based on bioluminescence or high content imaging (HCI) techniques have been used to carry out compound screening on gametocytes (Lelievre et al. Reference Lelievre, Almela, Lozano, Miguel, Franco, Leroy and Herreros2011), liver schizonts (Meister et al. Reference Meister, Plouffe, Kuhen, Bonamy, Wu, Barnes, Bopp, Borboa, Bright, Che, Cohen, Dharia, Gagaring, Gettayacamin, Gordon, Groessl, Kato, Lee, McNamara, Fidock, Nagle, Nam, Richmond, Roland, Rottmann, Zhou, Froissard, Glynne, Mazier, Sattabongkot, Schultz, Tuntland, Walker, Zhou, Chatterjee, Diagana and Winzeler2011; Derbyshire et al. Reference Derbyshire, Prudencio, Mota and Clardy2012) and hypnozoites (Dembele et al. Reference Dembele, Gego, Zeeman, Franetich, Silvie, Rametti, Le Grand, Dereuddre-Bosquet, Sauerwein, van Gemert, Vaillant, Thomas, Snounou, Kocken and Mazier2011). Although currently only low- to medium-throughput formats are available, these assays have been used to identify blood stage-active compounds with additional activity on other parasite life stages.
In our own efforts to discover new antimalarials, we screened both our chemical libraries including our unique natural product libraries for antimalarial chemotypes with activity on multiple plasmodium life stages. From this we identified two new antimalarial chemotypes; the spiroindolones, which display activity on sexual and asexual stages of the parasite and the imidazolopiperazines (IP), which are active on both sexual and asexual stages as well as liver schizonts. Over the past twenty years, these molecules are the first two antimalarial chemotypes with distinct and novel mechanisms of action to enter clinical development.
DISCOVERY AND OPTIMIZATION OF SPIROINDOLONE ANTIMALARIALS
Natural products-derived drugs are well represented in the current antimalarial arsenal (Wells, Reference Wells2011). Since the discovery of artemisinin from traditional Chinese medicine, derivatives of this natural product make up the current artemisinin-based combination therapies (ACTs). Coartem® was the first fixed dosed ACT approved by the WHO and since its release 2001, over 400 million treatments have been provided including a more recently developed paediatric formulation (http://www.coartem.com).
Coartem® remains highly efficacious but is under the threat of emerging artemisinin drug-resistance in South East Asia (Noedl, et al. Reference Noedl, Se, Schaecher, Smith, Socheat and Fukuda2008; Dondorp et al. Reference Dondorp, Nosten, Yi, Das, Phyo, Tarning, Lwin, Ariey, Hanpithakpong, Lee, Ringwald, Silamut, Imwong, Chotivanich, Lim, Herdman, An, Yeung, Singhasivanon, Day, Lindegardh, Socheat and White2009; Maude et al. Reference Maude, Pontavornpinyo, Saralamba, Aguas, Yeung, Dondorp, Day, White and White2009; Cheeseman et al. Reference Cheeseman, Miller, Nair, Nkhoma, Tan, Tan, Al Saai, Phyo, Moo, Lwin, McGready, Ashley, Imwong, Stepniewska, Yi, Dondorp, Mayxay, Newton, White, Nosten, Ferdig and Anderson2012; Phyo et al. Reference Phyo, Nkhoma, Stepniewska, Ashley, Nair, McGready, ler Moo, Al-Saai, Dondorp, Lwin, Singhasivanon, Day, White, Anderson and Nosten2012). Novel antimalarial drugs are thus urgently needed to anticipate this threat. In our first HTS campaign to find new antimalarial chemotypes, we used the Novartis natural product library which at the time contained, about 12000 pure natural products and natural product-like (synthetic) compounds to screen on the asexual blood stages of Plasmodium falciparum. From this we identified 275 primary hits which showed greater than 50% inhibition at less than 1·25 μ m (unpublished data). Removing known antimalarial scaffolds and reconfirming the remaining scaffolds in a [3H]-hypoxanthine uptake assay (Huber and Koella, Reference Huber and Koella1993) further refined the list of compounds. We also assessed cytotoxicity on a panel of mammalian cell lines and screened for cross resistance on chloroquine-resistant parasites (K1 strain). Because of the need for a tablet formulation, we were interested in identifying chemical scaffolds with some inherent oral bioavailability, and so we evaluated hit compounds in mouse ‘snapshot’ pharmacokinetic (PK) experiments (Liu et al. Reference Liu, Chang, Gordon, Isbell, Zhou and Tuntland2007). Other criteria utilized to help prioritize compounds included in vitro potency, where IC50 values were less than 1 μ m against wild type as well as drug-resistant strains; a favourable safety index, greater than 10 fold selectivity in a mammalian cell-line toxicity panel; and ease of synthesis or re-isolation of the natural product, to facilitate retesting or additional compound safety profiling (Plouffe et al. Reference Plouffe, Brinker, McNamara, Henson, Kato, Kuhen, Nagle, Adrian, Matzen, Anderson, Nam, Gray, Chatterjee, Janes, Yan, Trager, Caldwell, Schultz, Zhou and Winzeler2008). Not surprisingly we found that more than half of the natural product hits tested showed poor oral bioavailability, but out of the compounds identified which had some level of oral exposure in mice, one compound was chosen for further profiling. Although not the most potent hit, it displayed the highest oral exposure and displayed favourable physicochemical properties (aq. solubility: >75 μ m at pH 6·8, logP: 3·9).
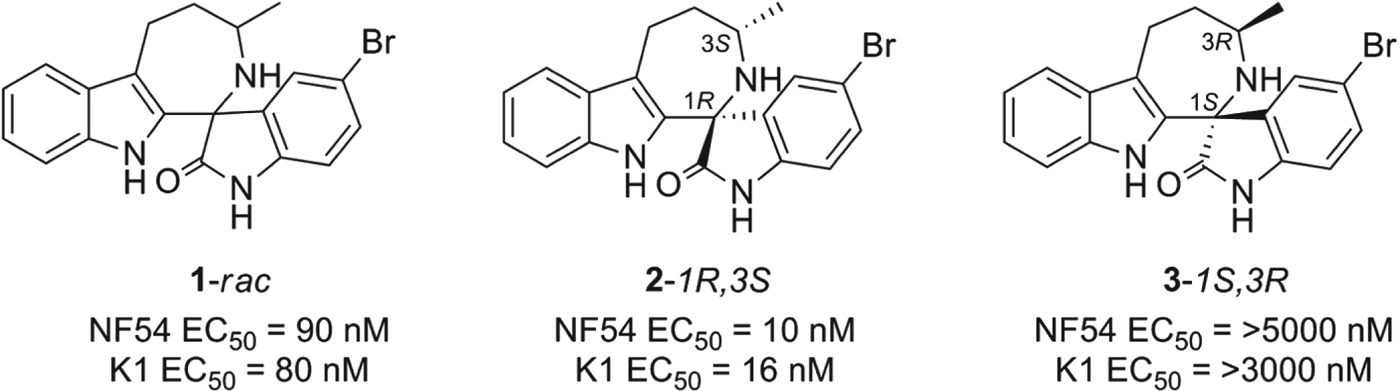
Interestingly the hit, compound 1 (Fig. 1), was not a natural product but had been purchased from a commercial source and added to the collection. The compound was a racemic mixture of unknown configuration and displayed moderate potency with both the drug-susceptible NF54 and chloroquine-resistant K1 P. falciparum strain (Yeung et al. Reference Yeung, Zou, Rottmann, Lakshminarayana, Ang, Leong, Tan, Wong, Keller-Maerki, Fischli, Goh, Schmitt, Krastel, Francotte, Kuhen, Plouffe, Henson, Wagner, Winzeler, Petersen, Brun, Dartois, Diagana and Keller2010). The central spirocentre imparted a unique overall shape when compared to the achiral, planar molecules often found in HTS screens and further distinguished itself from the other natural product hits which were structurally more complex. The compound displayed favourable physicochemical properties and offered a good starting point for a chemistry effort. In addition, the in vitro PK profile correlated well with a full in vivo PK experiment. In mice, 1 displayed good oral bioavailability (F=59%), and an oral half-life of nearly 4 h. Compound 1 also showed low cytotoxicity against a panel of mammalian cell lines, low risk for cardiotoxicity and low potential for drug-drug interactions (Yeung et al. Reference Yeung, Zou, Rottmann, Lakshminarayana, Ang, Leong, Tan, Wong, Keller-Maerki, Fischli, Goh, Schmitt, Krastel, Francotte, Kuhen, Plouffe, Henson, Wagner, Winzeler, Petersen, Brun, Dartois, Diagana and Keller2010).

Fig. 1. The antimalaria activity of the spiroindolone hit and its two enantiomers. The original hit (1) consisted of a mixture of enantiomers. Chiral chromatography followed by X-ray analysis was used to elucidate the absolute configuration of the enantiomeric mixture. The active enantiomer, 2, was determined to have the 1R,3S configuration.
One of the first tasks was to determine the exact configuration of both stereocentres. This was achieved by a chiral separation followed by an X-ray crystal structure. When the isomers were tested individually we observed a marked difference in potency of the two enantiomers 2 and 3, with only the 1R, 3S enantiomer responsible for activity on the parasite.
Compound 1 was evaluated in the Plasmodium berghei mouse model where remarkably, a single oral dose of 100 mg/kg resulted in a 96% reduction in parasitaemia. Although only a slight prolongation in mouse survival over control (seven days) was observed at this dose. We concluded that this was a highly promising starting point for optimization and that in vivo efficacy might be further improved upon increasing the potency of the compound.
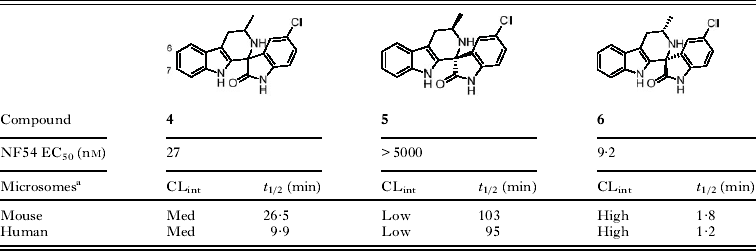
During the lead optimization of 1, we also prepared the six-membered spiro-derivatives exemplified by compound 4 and found that this change improved potency on the parasite by about three fold. Although chiral separation of the isomers confirmed the expected increase in potency upon isolating the active 1R,3S enantiomer over that of the racemic mixture, we observed additional differences in metabolic stability between enantiomers 5 and 6 (Table 1). In the presence of mouse and human liver microsomes, compound 4 displayed moderate metabolic stability (medium clearance), but when profiled separately, the active enantiomer (compound 6) was metabolized much more readily that the inactive enantiomer (compound 5). We postulated the indole ring system on compound 6 as metabolically susceptible and focused on introducing substituents to hinder oxidation of the indole ring.
Table 1. Differences between potency and metabolic stability of enantiomers
a Intrinsic clearance in liver microsomes; where high clearance corresponds to low stability in the presence of liver enzymes
The microsomal clearance assay proved to be a predictive tool in helping to assess the metabolic stability of the spiroindolones (Obach et al. Reference Obach, Baxter, Liston, Silber, Jones, MacIntyre, Rance and Wastall1997; Lau et al. Reference Lau, Krishna, Yumibe, Grotz, Sapidou, Norton, Chu, Chen, Soares and Lin2002). Derivatives which displayed high stability in vitro also had low clearance in vivo in mouse PK experiments. By systematically inserting fluorine substituents at the available positions on the indole moiety of 6, we found that blocking the C7 position had the greatest effect on increasing the half-life in the presence of liver microsomes (Table 2). As with C6, substitutions on positions C5 and C8 of the indole increased potency but did not improve the stability (data not shown). The 3- to 10-fold gains in potency were an added benefit with the 6,7-di-substituted derivatives having the most additive effects (compounds 9 and 10).
Table 2. Additive SAR of the spiroindolones where improving the metabolic stability also led to gains in potency

a Intrinsic clearance in liver microsomes; where high clearance corresponds to low stability in the presence of liver enzymes.
The spiroindolone optimizations described above translated remarkably well to efficacy in the P. berghei-infected mouse model. The spiroindolones 6, 9 and 10 were found to outperform chloroquine and artesunate under similar dosing regimens and experimental conditions (Table 3). When dosed orally, a single treatment of spiroindolones at 30 mg/kg displayed survival prolongation in mice greater than standard antimalarial controls and extended survival from 10 days to almost two weeks. Dosing with the compound once daily for three days (3×30 mg/kg) yielded more striking results with mouse survival out to 18·8 and 23·8 days with compounds 6 and 9, respectively. Interestingly, despite the high in vivo clearance and shortened half-life of 6, the compound achieved a 60% cure rate. Neither chloroquine nor artesunate was able to cure any mice under similar experimental conditions.
Table 3. In vivo efficacy of the optimized spiroindolonesa
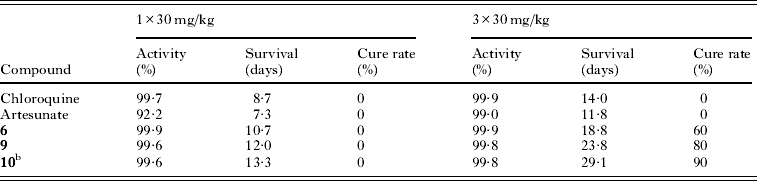
a Average parasitaemia reduction; survival of 6–7 days for untreated mice; cure was no parasites at day 30; compounds were formulated in 10% ethanol, 30% PEG400, 6% vitamin E TPGS;
b Formulated in 0·5% MCM/0·1% Solutol HS15.
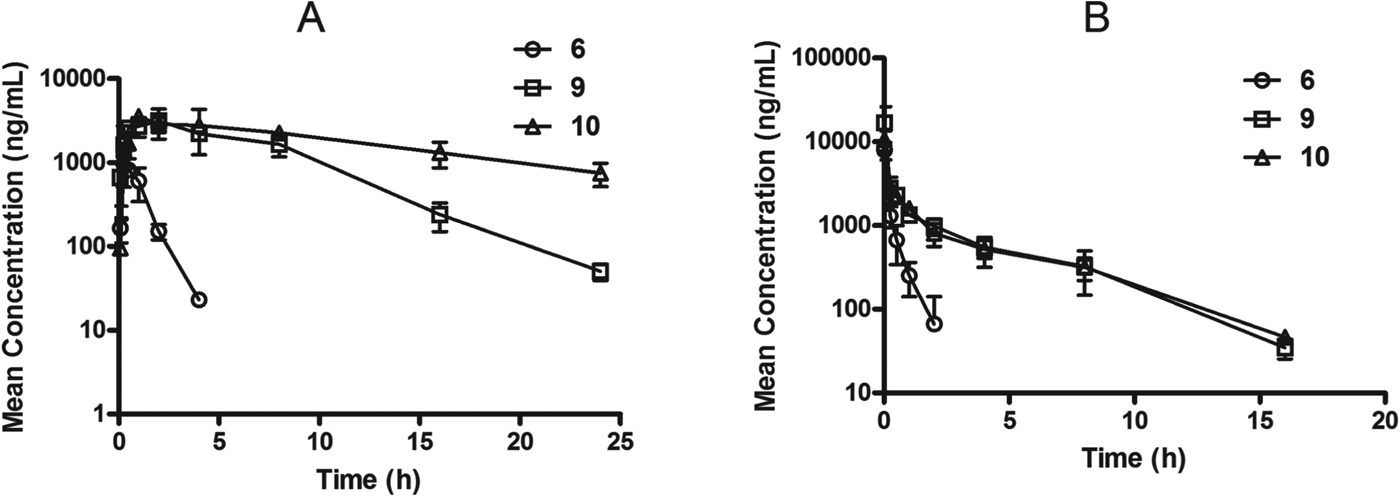
The in vitro PK profile for the spiroindolones tracked well with what was observed in vivo. The poor stability of compound 6 in liver microsomes was reflected in the in vivo mouse PK as was the improved stability of 9 and 10 (Fig. 2). Interestingly, despite the short half-life even compound 6 was able to achieve partial cure (60%) in the P. berghei mouse model at three daily doses (Table 3), while none of the standard antimalarial drugs displayed similar activity under these conditions. Compound 10 (NITD609) displayed the optimal combination of potency and oral exposure which warranted further preclinical evaluation.
Fig. 2. The plasma concentration versus time profile for spiroindolones 6 (○), 9 (□) and 10 (△) when administered orally (A) and intravenously (B) to mice. Plasma concentrations are indicated in ng/mL plus or minus standard deviations (n=3). The overall in vivo profiles correlate well with the in vitro microsomal clearance data.
Activity on gametocytes
As part of the eradication agenda identifying compounds that can not only clear a blood-stage infection but can also block the transmission of the parasite from human host to mosquito vector are needed. This is because asymptomatic carriers of P. falciparum gametocytes constitute a constant parasite reservoir in areas of high transmission. New antimalarial drug candidates are now routinely evaluated for transmission-blocking activity (Delves et al. Reference Delves, Plouffe, Scheurer, Meister, Wittlin, Winzeler, Sinden and Leroy2012) and compound 10 was tested on early and late stage gametocytes as well as in the standard membrane feeding assay (SMFA) to test the potential effects of the drug on sporogonic development in mosquitoes (van der Kolk et al. Reference van der Kolk, De Vlas, Saul, van de Vegte-Bolmer, Eling and Sauerwein2005). Consistent with its sub-nanomolar potency for asexual stages, NITD609 displayed dose-dependent activity on early (stage II) and late gametocyte (stage V) development assays from 5 to 500 nm, with very significant gametocytaemia reduction at the lowest tested concentration of 5 nm. NITD609 also shows potent activity in the SMFA and completely blocks transmission of the parasite at 500 nm, suggesting a further inhibiting effect on the stage V mature gametocytes taken up by the mosquito and entering fertilization in the mosquito midgut and/or on the production of ookinetes or oocysts (van Pelt-Koops et al. Reference van Pelt-Koops, Pett, Graumans, van der Vegte-Bolmer, van Gemert, Rottmann, Yeung, Diagana and Sauerwein2012). These data highlight the compound as a potential transmission-blocking drug with inhibitory activity on more than one critical step in the sexual stages of the parasite life cycle.
The target of the spiroindolones
The application of drug pressure to cultured parasite clones can both assess the potential for drug resistance evolution as well as provide insight into the mechanism of action. We used this method to evolve resistant mutants and compare changes in their DNA to wild type parasites (Rottmann et al.). After three to four months of exposing P. falciparum parasites to sub-lethal doses of NITD609, we observed IC50 shifts between 7- to 24-fold (3 to 11 nm). The low level of resistance observed suggested that the spiroindolones do not readily select for a high degree of resistance in vitro. In addition, none of the spiroindolone resistant mutants showed cross-resistance to artemisinin or mefloquine. Genomic DNA (gDNA) was prepared from drug-resistant clones then fragmented, labelled, and hybridized to a high density tiling array. The differences between the gDNA of wild type and spiroindolone resistant clones were compared and detectable as single nucleotide polymorphisms (SNPs) or copy number variations (CNP) which mapped to a single gene, pfatp4 that encodes a cation-transporting P-type adenosine triphosphatase (ATPase) (PfATP4) (Krishna et al. Reference Krishna, Woodrow, Webb, Penny, Takeyasu, Kimura and East2001). This family of ATP-dependent transporters are inhibited by a diverse set of compounds, including thapsigargin, cyclopiazonic acid and lansoprazole, and thereby constitutes an attractive set of drug targets (Yatime et al. Reference Yatime, Buch-Pedersen, Musgaard, Morth, Lund Winther, Pedersen, Olesen, Andersen, Vilsen, Schiøtt, Palmgren, Møller, Nissen and Fedosova2009). P-type ATPases are ubiquitous in eukaryotic organisms, and those involved in divalent cation transport appear to maintain a conserved structural mechanism for ion translocation (Kühlbrandt, Reference Kühlbrandt2004). Recent experiments have shown that PfATP4 is a key regulator of intracellular Na+ homeostasis for the parasite and that spiroindolones potently interfere with this physiological process. Exposure to compound 9 causes a rapid and sharp increase Na+ concentration and an alkalinisation of the Plasmodium parasite cytosol (Spillman et al. Reference Spillman, Allen, McNamara, Yeung, Winzeler, Diagana and Kirk2013). These data provide further evidence that PFATP4 is likely to be the drug target of the spiroindolones.
Retrospectively, compound 1 represented a high quality hit with favourable physicochemical properties and good oral PK properties. The spirocentre imparted a three dimensionality to the overall shape of the molecule which we believe gave the molecule good intrinsic solubility (100 μ m at pH 6·8) not generally encountered in highly planar, aromatic compounds. Although part of our natural product collection, compound 1 is not a natural product and instead is fully synthetic (Grigoryan et al. Reference Grigoryan, Pogosyan and Paronikyan2005). Finally, compound 1 also happened to represent the minimum pharmacophore of the series. Nearly all of the essential structural components required for potency in the optimized compound were already present in the hit (e.g. the spirocentre, 3-methyl substituent, and the substituted oxindole moiety). We believe this is another one of the key reasons we were able to optimize the series rapidly and efficiently. NITD609 (10) has completed Phase I safety studies in man and is currently in Phase II clinical trials.
DISCOVERY AND OPTIMIZATION OF IMIDAZOLOPIPERAZINE ANTIMALARIALS
The imidazolopiperazines represent the second scaffold from our portfolio to achieve preclinical development status. From a library of 5697 blood stage-active compounds, 275 were confirmed as active against liver stage parasites (Wu et al. Reference Wu, Nagle, Kuhen, Gagaring, Borboa, Francek, Chen, Plouffe, Goh, Lakshminarayana, Wu, Ang, Zeng, Kang, Tan, Tan, Ye, Lin, Caldwell, Ek, Skolnik, Liu, Wang, Chang, Li, Hollenbeck, Tuntland, Isbell, Fischli, Brun, Rottmann, Dartois, Keller, Diagana, Winzeler, Glynne, Tully and Chatterjee2011); with 229 compounds having an IC50 of less than 1 μ m, and 86 compounds less than 100 nm. Additionally, this dataset of 275 active compounds contained chemical scaffolds distinct from known antimalarial pharmacophores (quinolines, quinones and trioxanes) and devoid of reactive functional groups (such as Michael acceptors or nitroarenes). Clustering the scaffolds identified a chemically tractable series of imidazolopiperazines structurally unrelated to any known antimalarial drug class (Fig. 3). Moreover, the series displayed no cross-resistance against parasite strains resistant to chloroquine and/or pyrimethamine, implying a potentially distinct mechanism of action.
Fig. 3. The initial hit of imidazolopiperazine series (11a) and the strategy to optimize the three peripheral parts.
The original hits (compounds 11a–c) were part of the imidazolopiperazines (IP) class and represented an attractive series not only for their dual-stage activity (Meister et al. Reference Meister, Plouffe, Kuhen, Bonamy, Wu, Barnes, Bopp, Borboa, Bright, Che, Cohen, Dharia, Gagaring, Gettayacamin, Gordon, Groessl, Kato, Lee, McNamara, Fidock, Nagle, Nam, Richmond, Roland, Rottmann, Zhou, Froissard, Glynne, Mazier, Sattabongkot, Schultz, Tuntland, Walker, Zhou, Chatterjee, Diagana and Winzeler2011) but potency on both drug-susceptible and -resistant parasite strains while maintaining an acceptable cytotoxicity index on Huh7 cells (Wu et al. Reference Wu, Nagle, Kuhen, Gagaring, Borboa, Francek, Chen, Plouffe, Goh, Lakshminarayana, Wu, Ang, Zeng, Kang, Tan, Tan, Ye, Lin, Caldwell, Ek, Skolnik, Liu, Wang, Chang, Li, Hollenbeck, Tuntland, Isbell, Fischli, Brun, Rottmann, Dartois, Keller, Diagana, Winzeler, Glynne, Tully and Chatterjee2011) (Table 4).
Table 4. The antimalarial activity and cytotoxicity of the imidazolopiperazine hits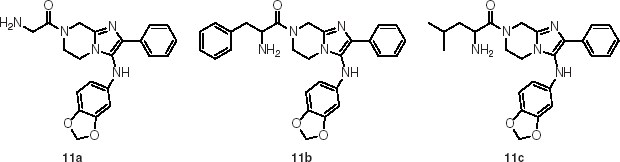

a Values are mean of two experiments.
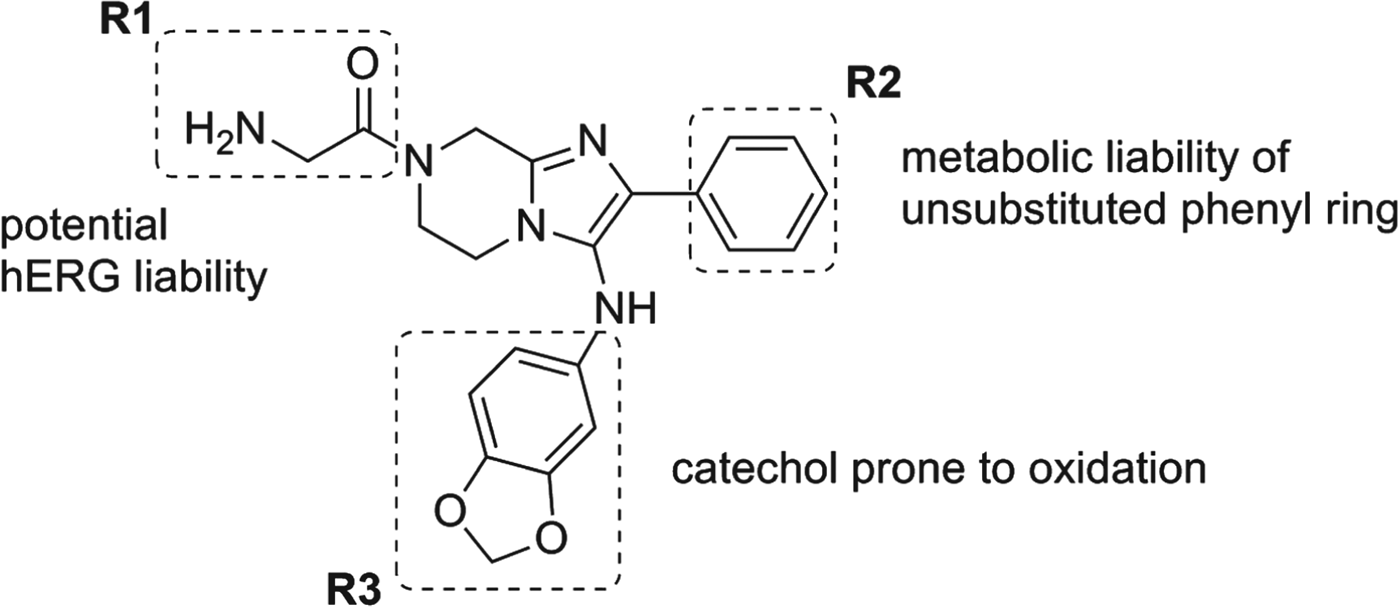
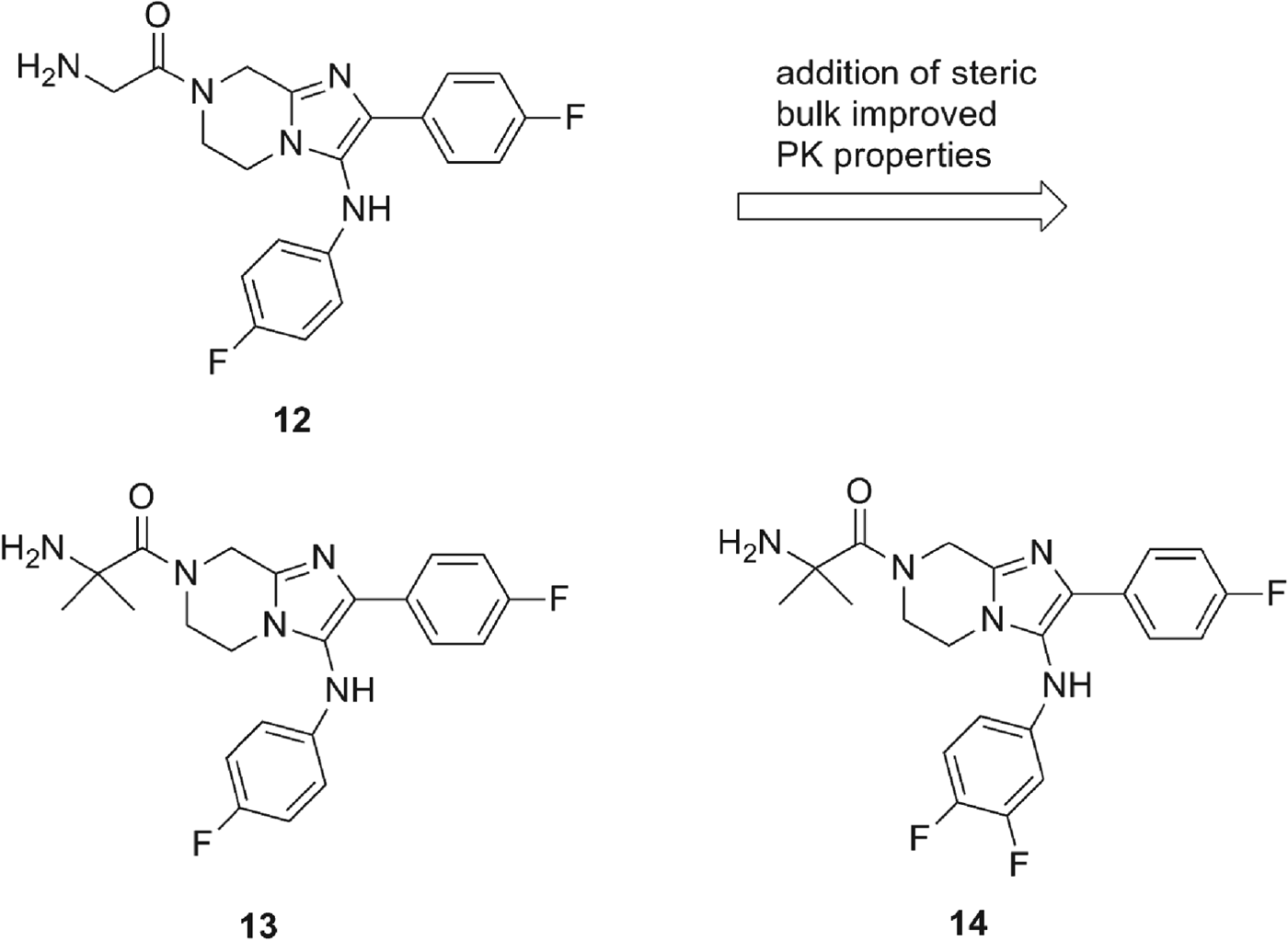
The progression of this series was facilitated by several key observations during the early development of the structure/activity relationship (SAR). The application of in silico tools identified potential metabolic liabilities of the scaffold which allowed us rapidly to address them chemically without microsomal stability data. This type of approach allowed us to replace the dioxalane which was known to be prone to enzymatic oxidation and address the unsubstituted phenyl ring at the 2-position as another potential area of metabolic oxidation. Of the original hits, compound 11a was highly soluble (>175 μ m at pH 6·8) and clean against most commonly drug-affected cytochrome P450 isoforms (IC50>10 μ m). However, a moderate hERG signal was detected in the binding assay (IC50=19 μ m, 57% inhibition at 10 μ m) (Dubin et al. Reference Dubin, Nasser, Rohrbacher, Hermans, Marrannes, Grantham, Van Rossem, Cik, Chaplan, Gallacher, Xu, Guia, Byrne and Mathes2005) which we attributed to the basic amine in the glycine moiety at R1 (Fig. 3). Despite the good solubility, compound 11a exhibited poor plasma oral exposure with C max=320 nm and AUC(0–5h)=972 (nm·h) in a mouse oral snapshot PK study, indicating the presence of metabolic soft spots within the molecule or overall poor permeability properties in a passive permeability (PAMPA) assay. The main objective of the optimization was to improve potency and remove metabolically vulnerable functionality of the compounds. We sought to achieve this by focusing on three peripheral regions of the molecule: the amino acid moiety R1, the phenyl moiety R2 and the aniline moiety R3; and we surmised that changes in these parts would steer us towards a candidate with enhanced potency as well as improved oral exposure (Fig. 3). In the first round of analogues, R2 was found to be intolerant of broad structural changes and hence the 4F-phenyl group was quickly identified as optimal. R3 substituents including aliphatic rings and heterocyclic rings were not tolerated. Only aniline derivatives bearing either meta- or para-substitutions produced active compounds. Another trend observed was that compounds incorporating glycine at R1 were generally more active when compared with other amino acid substitutions and that the primary amine was essential for activity (data not shown). A balance was struck with the α-methyl alanine derivatives where despite a marginal loss of potency, we observed an improved in vitro PK profile (Fig. 4, Table 5). We attributed the improved compound profile to the increase in microsomal stability while maintaining good physicochemical properties of the scaffold; however the hERG liability remained (Table 5). The α-methyl alanine derivatives exemplified by compounds 13 and 14 achieved about a six-fold increase in C max (1803 and 1940 nm respectively) and an 18 to 30-fold increase in oral AUC (17 894 and 31 758 nm·h, respectively) over compound 11a.
Fig. 4. Installing the α-methyl alanine moiety at R3 lead to improved potency over the hit compound and provided vast improvements in the PK profile of the compound (Tables 5 and 6).
Table 5. Comparison of the glycine and α-methyl-alanine derivatives
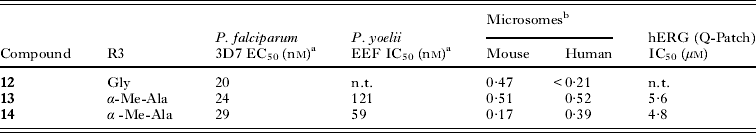
a Values are mean of two experiments;
b Hepatic stability in microsomes expressed as extraction ratio (Obach et al.); n.t.=not tested.
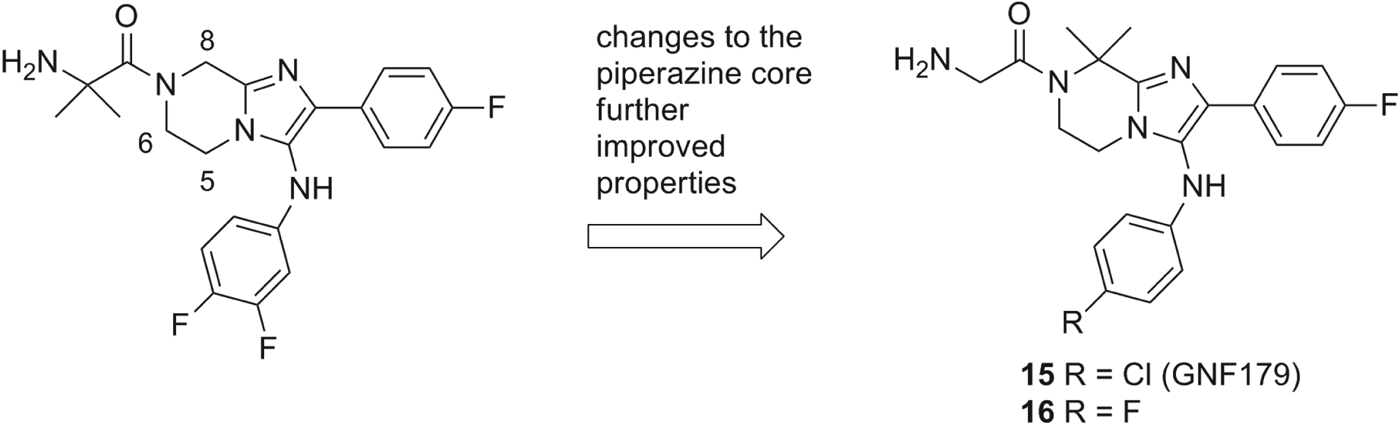
The next advance in the SAR came with analogues in which the gem-dimethyl moiety was shifted from R1 to the piperazine core (Fig. 5). We investigated various substitutions at positions 5 and 6 but it soon became apparent that position 8 was optimum. The gem-dimethyl group provided the most significant potency boost on liver stage EEFs while the similar replacement at position 5 or 6 did not (data not shown). Although the metabolic stability and overall physicochemical profiles remained unchanged from the previous series, we noticed an improvement in reducing the hERG liability (Table 6). hERG inhibition was reduced by at least 2- to 3-fold in the Patch clamp assay suggesting a lower potential for QT prolongation (Zheng et al. Reference Zheng, Spencer and Kiss2004; Mathes, Reference Mathes2006) and an improved early safety profile. Compounds 15 and 16 exhibited favourable PK in mice with a C max range (1200 and 1538 nm, respectively) and comparable AUC(0–24h) (20 700 and 12 155 nm·h, respectively) to previous compounds 13 and 14.
Fig. 5. Changes in the core piperazine ring were found further improve the activity in both blood and liver-stage assays. More specifically the 8,8-dimethyl imidazolopiperazine modification maintained a favorable oral exposure in mice which translated to improved efficacy in the malaria mouse model (compound 14 vs. 15). Potential for cardiotoxicity was also decreased with this modification.
Table 6. Representative examples of dimethyl piperazine analogues with improved potency and cardiotoxicity profile
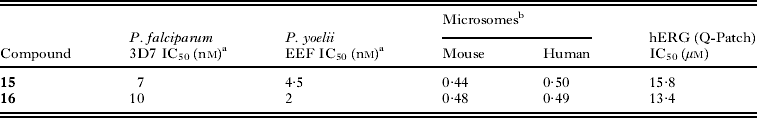
a Values are mean of two experiments; b Hepatic stability in microsomes expressed as extraction ratio (Obach et al.); n.t.=not tested.
Compounds from the imidazolopiperazine series with suitable physicochemical properties and activities were routinely tested in the P. berghei malaria mouse model and compounds 14 and 15 were tested to assess how the improved in vitro activity translated to in vivo efficacy. Both compounds were highly efficacious in vivo with >99% parasitaemia reduction and displayed comparable or slightly improved survival prolongation to that of chloroquine or artesunate under similar dosing regimens (Table 7).
Table 7. The in vivo efficacy of compounds 14 and 15 (GNF179) compared to standard antimalarial drugsa
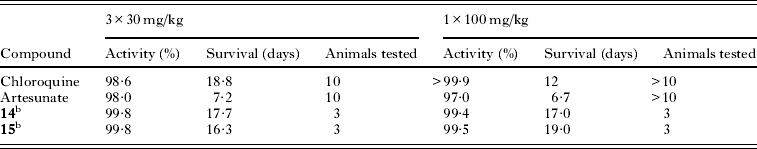
a Average parasitaemia reduction; survival of 6–7 days for untreated mice; cure was no parasites at day 30; compounds were formulated in 7% Tween80, 3% ethanol;
b Formulated in PEG300/D5W.
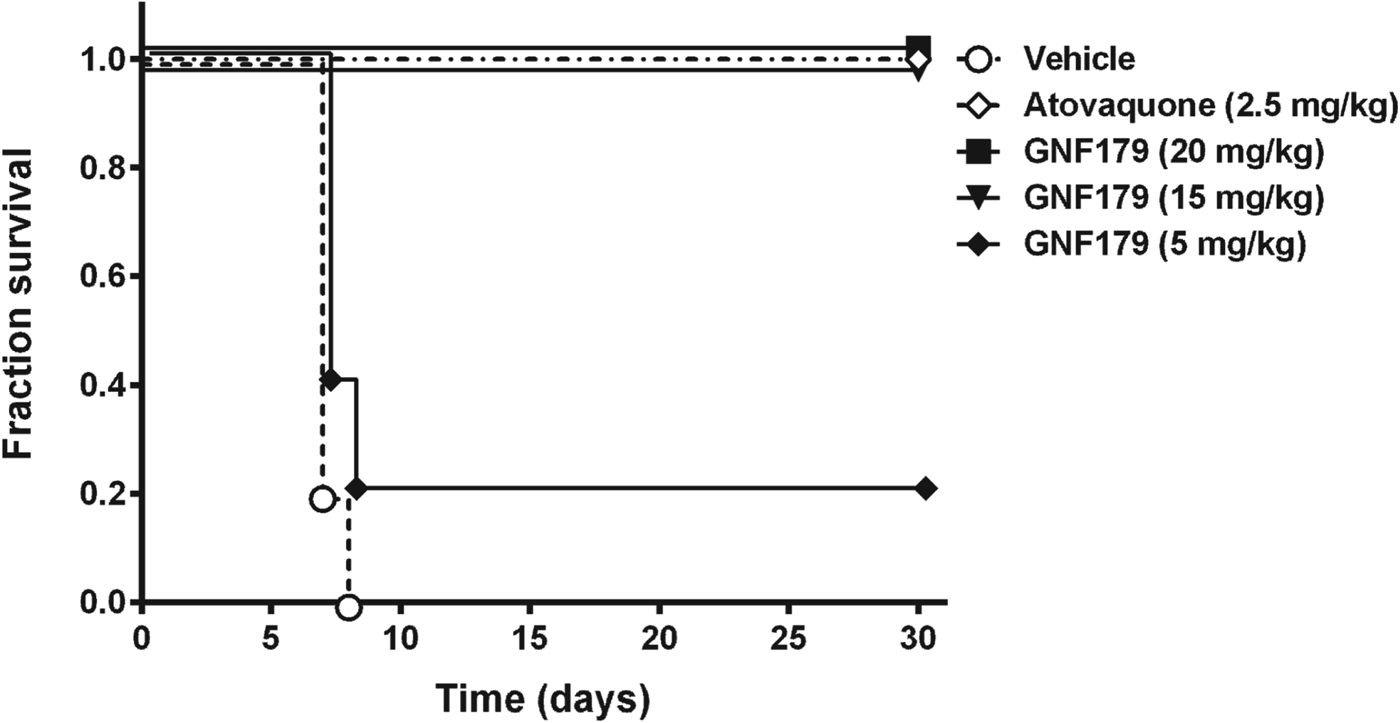
We used a rodent malaria model of causal prophylactic treatment to confirm the tissue schizonticidal activity observed in vitro with compound 15 (GNF179) (Fig. 6). Mice were given a single dose of either atovaquone (positive control) or GNF179 and then injected with 105P. berghei sporozoites. GNF179 was able to fully protect against a P. berghei challenge infection at a single dose of 15 mg/kg. This result correlated well with the in vitro activity on P. yoelii EEFs and we concluded that compounds identified using this cellular screening approach offer evidence of additional causal prophylactic activity.
Fig. 6. Compound 15 is fully protective in a causal prophylactic mouse model of malaria. Mice (n=5) were infected with P. berghei sporozoites (i.v.) on day 0 and administered a single dose of compound an hour later. The mice were monitored over 30 days for parasitemia. Vehicle used for compound administration was a suspension formulation of 0.5% w/v methylcellulose, 1% (w/v) solutol HS15. Data represents the number of live mice at the indicated day post-infection.
In total, over 1200 analogues of the imidazolopiperazine series were prepared by first optimizing their blood-stage potency then testing for liver-stage activity on P. yoelii EEFs. We generally observed that the in vitro activity on liver stages tracked well with efficacy in the rodent causal prophylaxis model where low nanomolar activity in vitro translated to full protection at low, single oral doses.
The target of the imidazolopiperazines
GNF179 is unlike other compounds which target early hepatic stages of the parasite and does not inhibit protein biosynthesis like cycloheximide, nor does it show cross resistance with atovaquone-resistant strains; precluding acting by inhibition of cytochrome bc1 (Meister et al. Reference Meister, Plouffe, Kuhen, Bonamy, Wu, Barnes, Bopp, Borboa, Bright, Che, Cohen, Dharia, Gagaring, Gettayacamin, Gordon, Groessl, Kato, Lee, McNamara, Fidock, Nagle, Nam, Richmond, Roland, Rottmann, Zhou, Froissard, Glynne, Mazier, Sattabongkot, Schultz, Tuntland, Walker, Zhou, Chatterjee, Diagana and Winzeler2011; Nam et al. Reference Nam, McNamara, Bopp, Dharia, Meister, Bonamy, Plouffe, Kato, McCormack, Bursulaya, Ke, Vaidya, Schultz and Winzeler2011). Applying a similar methodology to that used to develop spiroindolone resistance, mutants were evolved under sub-lethal drug pressure and cloned. Their genomes were compared to wild-type parasites using high-density microarrays or full genome sequencing where a single gene (PFC0970w), termed P. falciparum cyclic amine locus (pfcarl), was found to be mutated in all strains. No other genetic changes were found in any other known P. falciparum-resistant genes including pfmdr1 or pfcrt. The pfcarl gene encodes for an uncharacterized protein with seven predicted transmembrane domains. Homologues of pfcarl are found across all eukaryotes including humans and its homologue in Caenorhabditis elegans is essential (Maeda et al. Reference Maeda, Kohara, Yamamoto and Sugimoto2001). Previous work with yeast deletions strains (Winzeler et al. Reference Winzeler, Shoemaker, Astromoff, Liang, Anderson, Andre, Bangham, Benito, Boeke, Bussey, Chu, Connelly, Davis, Dietrich, Dow, El Bakkoury, Foury, Friend, Gentalen, Giaever, Hegemann, Jones, Laub, Liao, Liebundguth, Lockhart, Lucau-Danila, Lussier, M'Rabet, Menard, Mittmann, Pai, Rebischung, Revuelta, Riles, Roberts, Ross-MacDonald, Scherens, Snyder, Sookhai-Mahadeo, Storms, Veronneau, Voet, Volckaert, Ward, Wysocki, Yen, Yu, Zimmermann, Philippsen, Johnston and Davis1999) suggests that pfcarl may play a role in protein folding within the endoplasmic reticulum and is presumably required for both hepatic and blood stages (Jonikas et al. Reference Jonikas, Collins, Denic, Oh, Quan, Schmid, Weibezahn, Schwappach, Walter, Weissman and Schuldiner2009). Although additional studies are needed, these preliminary data suggest that the IPs work through a distinct mechanism of action different from known antimalarial drugs.
TARGETING PLASMODIUM VIVAX LIVER STAGES FOR NEW RADICAL CURE AGENTS
The two antimalarial compounds described above were the product of phenotypic screening approaches on the asexual blood stages of P. falciparum which to date is the only human malaria parasite that can be cultured in vitro. Presently, the determination of antimalarial activity against P. vivax (blood stages) relies on ex vivo culture systems (Price et al. Reference Price, Tjitra, Guerra, Yeung, White and Anstey2007; Malleret et al. Reference Malleret, Claser, Ong, Suwanarusk, Sriprawat, Howland, Russell, Nosten and Renia2011; Russell et al. Reference Russell, Suwanarusk, Malleret, Costa, Snounou, Baird, Nosten and Rénia2012) and access to the dormant hypnozoite liver stages is challenging and does not currently allow for routine compound screening (Chattopadhyay et al. Reference Chattopadhyay, Velmurugan, Chakiath, Andrews Donkor, Milhous, Barnwell, Collins and Hoffman2010). In P. vivax, invading parasites entering the liver have two fates. Some of the parasites continue on to develop into schizonts and ultimately merosomes that will yield the blood stage-acute and -symptomatic infection, while an unknown and variable subset of parasites develops into dormant hypnozoites causing a chronic infection and disease relapses (Cogswell, Reference Cogswell1992). The development of a robust P. vivax liver-stage culture system is critical for accessing the hypnozoite stage and would enable the screening of chemical libraries to identify new classes of P. vivax radical cure drugs with different modes of action from primaquine (8-aminoquinolines). Since access to P. vivax parasites remains limited to clinical isolates, there remains a need for the use of surrogate Plasmodium species (i.e. P. yoelii, P. berghei, and P. cynomolgi).
Besides the lack of reliable access to parasite liver stages, the search for new radical cure agents has also been hampered largely from the dearth of biological tools available for working with P. vivax parasites. P. cynomolgi, a simian malaria which shares the same ability as P. vivax to form dormant hypnozoites, has been used as the gold standard in an infected rhesus monkey model. The discovery of primaquine was carried out using this model but for ethical reasons and prohibitive costs, rhesus monkeys cannot be used to screen potential anti-relapse drugs. Recently however, there have been efforts to develop a predictive assay based on this in vivo model which has lead to a method of establishing P. cynomolgi infections of hepatocytes in vitro (Dembele et al. Reference Dembele, Gego, Zeeman, Franetich, Silvie, Rametti, Le Grand, Dereuddre-Bosquet, Sauerwein, van Gemert, Vaillant, Thomas, Snounou, Kocken and Mazier2011). In this assay P. cynomolgi sporozoites were used to infect primary monkey hepatocytes whereby, on infection, two distinct hepatic forms of the parasite were observed, large mature schizonts and a uninucleate small form reminiscent of hypnozoites. Moreover, the two liver forms were found to have different dose-dependent drug susceptibilities in the presence of primaquine and atovaquone. Atovaquone mainly affected large schizonts while primaquine cleared both schizonts and hypnozoite forms. The different drug-sensitivity profiles of liver stage parasites are consistent with the known biological activities of the two drugs; namely causal prophylactic activity for atovaquone and both causal prophylactic activity and radical cure for primaquine. Although currently low throughput, the assay allows, for the first time, in vitro compound testing on both liver stages of a P. cynomolgi infection.
Clearly a number of challenges remain before the HTS screening of chemical libraries against P. cynomolgi liver stages becomes realized. If successful, the P. cynomolgi liver-stage assay would be a powerful new tool to identify compounds with potential radical curative activity. When used in concert with existing assays that target other parasite life stages it will be possible to identify the next generation antimalarials with activity on more than one parasite life stage.
Although the antimalarial drug pipeline has been bolstered in recent years with the addition of new chemical entities in preclinical and clinical development, the relative rise of P. vivax malaria burden has the potential to hinder malaria control programmes if the newly introduced classes of drugs do not effectively kill P. vivax hypnozoites and block disease transmission. A paradigm change from the drug discovery approach which previously only focused on asexual blood stages has shifted to one where new drugs are expected to target multiple life stages of the parasite. Compounds with activity in multiple parasite assays can be prioritized for hit evaluation and lead optimization (Fig. 7). When combined with reverse genetic techniques, these chemical scaffolds can be used potentially to elucidate new drug targets or MOAs. Both the spiroindolones and inidazolopiperazines were the product of a high-throughput phenotypic screening campaign and represent the successful implementation of this multiple assay approach.
ACKNOWLEDGEMENTS
We gratefully acknowledge the individual members of the NGBS consortium whose work is summarized here.
FINANCIAL SUPPORT
The work was supported by a translational research grant (WT078285 and WT096157) to the Genomics Institute of the Novartis Research Foundation, the Biomedical Primate Research Center, the Swiss Tropical and Public Health Institute, and the Novartis Institute for the Tropical Diseases from the UK. Wellcome Trust and Medicines for Malaria Venture.