I. INTRODUCTION
The need to develop new, clean, and efficient synthesis is on demand nowadays, in particular those associated with the direct mixture of reactants without a solvent because they avoid unwanted contaminants, making these methodologies less harmful to the ambient. The solvent-free preparation of products of different natures has been reported (Tanaka, Reference Tanaka2003); in the case of the formation of C-N bonds, Wenz et al. (Reference Wenz, Steinbrunn and Landfester1997) prepared polyamides from α, ω–amino acids contained as inclusion compounds in α-cyclodextrin by the heating of the microcrystalline powder at temperatures above 473 K. Recently, Vouyiouka et al. (Reference Vouyiouka, Karakatsani and Papaspyride2005) proposed kinetic models to explain polyamide solid-state polymerization of nylon 6.6 resins by heating at temperatures of 433 to 473 K. Furthermore, the condensation product of the direct mixture of carboxylic acids and primary amines is amides; reactions of this type have been recently carried out using microwave irradiation (Perreux et al., Reference Perreux, Loupy and Volatron2002). On the basis of these works, we proposed the direct conversion of N-carbamoyls to hydantoins by heating of reagent at certain temperature. Hydatoins or imidazolidine-2,4-diones are compounds with an imidazolic ring that have keto groups in positions 2 and 4. Depending on the nature and type of substitution on the heterocyclic ring, these compounds may display pharmaceutical and biological activity with a variety of applications: fungicidal (Marton et al., Reference Marton, Enisz, Hosztafi and Timar1993), antiviral (Opačić et al., Reference Opačić, Barbarić, Zorc, Cetina, Nagl, Frković, Kralj, Pavelić, Balzarini, Andrei, Snoeck, De Clercq, Raić-Malić and Mintas2005), herbicide (Hanessian et al., Reference Hanessian, Sanceau and Chemla1995), antitumoral and anti-inflammatory (Thenmozhiyal et al., Reference Thenmozhiyal, Wong and Chui2004), protein inhibitor (Wu et al., Reference Wu, Emeigh, Gao, Goldberg, Kuzmich, Miao, Potocki, Qian, Sorcek, Jeanfavre, Kishimoto, Mainolfi, Nabozny, Peng, Reilly, Rothlein, Sellati, Woska, Chen, Gunn, O’Brien, Norris and Kelly2004), HIV protease inhibitor (Comber et al., Reference Comber, Reynolds, Friedrich, Manguikian, Buckheit, Truss, Shannon and Secrist1992), hypolipidimic agent (Tompkins, Reference Tompkins1986), and antihypertensive agent (Kieć-Kononowicz et al., Reference Kieć-Kononowicz, Stadnicka, Mitka, Pękala, Filipek, Sapa and Zygmunt2003). However, hydantoins are better known as anticonvulsants (Merrit and Putnam, Reference Merrit and Putnam1938); if they are substituted by aliphatic groups they display hypnotic properties, while those substituted by phenyl groups, such as diphenylhydantoin, display antiepileptic activity (Camerman and Camerman, Reference Camerman and Camerman1971; Thenmozhiyal et al., Reference Thenmozhiyal, Wong and Chui2004). Poupaert et al. (Reference Poupaert, Vandervorst, Guiot, Mustafa and Dumont1984) and Cortes et al. (Reference Cortes, Liao, Watson and Kohn1985) observed a reduction on the anticonvulsant activity in 5,5-diphenylhydantoin when the imidazolidin-2,4-dione ring was compared with analogous compounds with imidazol-2-one and 2-thioxoimidazolidin-4-one rings. The reduction of the biological activity was attributed to alteration of the hydrogen donor and acceptor capacity of the heterocyclic rings. Recently, the biocatalytic conversion of hidantoins has called the attention since it is a source for production of natural and non-natural amino acids. The reaction involves two consecutive hydrolysis catalyzed by two enzymes: (1) dihydropyrimidase (EC 3.5.2.2), which converts 5-monosubstituted hydantoins into L- or D-N-carbamoyls; (2) N-carbamoylase (EC 3.5.1), which converts the L- or D-N-carbamoyls into L- or D-α–amino acids (Burton and Dorrington, Reference Burton and Dorrington2004).
Here, we report the conversion of N-carbamoyl-L-proline to hydantoin-L-proline by direct heating at 470 K. The melt product was allowed to solidify and the crystal structure of hydantoin-L-proline was solved and refined from powder synchrotron diffraction data.
II. EXPERIMENTAL
A. Synthesis
Hydantoin-L-proline (II) was obtained from N-carbamoyl-L-proline (I) by the solvent-free reaction described

Figure 1. (Color online) (a) TGA plot of N-carbamoyl-L-proline; (b) DSC plot of N-carbamoyl-L-proline.
here. The N-carbamoyl (I) (mp of 476 to 478 K) was obtained from L-proline following a synthetic procedure described elsewhere (Seijas et al., Reference Seijas, Delgado, Mora, Bahsas and Uzcategui2006). FT-IR has 1695.5 cm−1 [t, C=O (acid group)] and 1660.8 cm−1 [t, C=O (carbamyl group)]. 1H NMR has (400 MHz, DMSO-d6) δ=12.45 (b-s, H2), 5.88 (s, H2A, H2B), 4.14 (dd, H4), 3.32 (m, H1B), 3.23 (m, H1A), 1.84 (m, H3A), 2.06 (m, H3B), and 1.84 (H2C and H2D). 13C NMR has (100.6 MHz, DMSO-d6) δ=7 (C5), 157.2 (C6), 58.4 (C4), 46.4 (C1), 29.6 (C3), and 24.4 (C2). 200 mg (1.266 mmol) of (I) was placed on a glass reactor under reduce pressure; the reactor was heated at a rate of 5°/min from room temperature to 476 K, keeping the reactor at this temperature for 10 min until the complete fusion of the solid. The melt was allowed to cool to room temperature observing a change in the color of the solid from white to beige (yielding 98.0 mg, 0.700 mmol, and mp of 457 to 458 K). FT-IR has (in KBr Pallets) 3443 cm−1 (t, N-H), 1757 cm−1 (tsym, C=O), 1708 cm−1 (tasym, C=O), and 1405 cm−1 (t, C-N). 1H NMR has (400 MHz, DMSO-d6) δ=10.4 (H3, b-s), 4.07 (H5A, t, J1=4 Hz), 3.43 (H8B, m), 3.02 (H8A, m), 3.23 (H1A, m), 1.95 (H7A=H7B m), 2.03 (H6B, m), and 1.60 (H6A, m); 13C NMR has (100.6 MHz, DMSO-d6) δ=161.0 (C2), 175.4 (C4), 64.0 (C5), 44.9 (C8), 26.7 (C6), and 26.6 (C7). Additional signals in the 1H NMR with very low intensity were observed at 1.75, 3.25, and 3.75 ppm.
B. Thermal analysis
For the thermogravimetric analysis, sufficient quantities of N-carbamoyl-L-proline (I) (4.1 mg) and hydantoin-L-proline (II) (5.3 mg) were placed on the aluminum canisters of a thermobalance Perkin-Elmer TGA7 programmed to heat the samples from 295 to 623 K at a rate of 10 K min−1 under a dry N2 flux of 50 mL min−1. The thermograms for (I) were recorded and are shown in Figure 1.
C. Synchrotron X-ray powder diffraction
X-ray powder diffraction data of (I) and (II) were collected with the high-resolution X-ray powder diffractometer on beamline ID31, ESRF (Fitch, Reference Fitch2004), selecting X-rays from an undulator source with wavelengths of 0.799 93(1) Ǻ. Small quantities of (I) and (II) were lightly ground with a pestle in an agate mortar and introduced into 1.0-mm-diameter borosilicate glass capillaries, mounted on the axis of the diffractometer and spun at approximately 1 Hz during measurements. Data were collected in continuous mode, normalized against monitor counts and detector efficiencies, and rebinned into steps of 2θ=0.003°.
III. RESULTS AND DISCUSSION
Figure 1a shows the TGA plot of (I). A weight loss of 10.6% occurs around 470 to 473 K, which can be explain as shown in Figure 2: (a) loss of a H2O molecule, corresponding to the formation of tetrahydro-pyrrol-[1,2-c]-imidazol-1,3-dione or hydantoin-L-proline (IIa); (b) loss of a NH3 molecule, which will produce the tetrahydro-pyrrol-[1,2-c]-oxazol-1,3-dione (IIb); or (c) both thermal reactions occurs simultaneously. Additional decomposition peaks are also seen at 489 and 530 K. The DSC plot [Figure 1b] of (I) shows the melting and decomposition peaks overlapping at 470.7 K. Also, the baseline of the DSC rises prior to melting, which is an evidence of the onset of the thermal reaction IIa

Figure 2. Two possible reaction routes for the thermal decomposition of N-carbomoyl-L-proline (i) at 473 K. Cyclization of (i) to form an imidazol ring (IIa) and water (left). Cyclization of (i) to form an oxazol ring (IIb) and ammonia (right). Both reactions explain a weight loss of around 10±2 % in the TGA experiment.

Figure 3. (Color online) 1H-1H COSY spectra of the reaction product.
or IIb, which changes the nature of the compound and hence its molar heat capacity (C p). The 1H NMR spectra of the thermal reaction product show all the signals corresponding to compound IIa and few additional peaks of very low intensity, which could be assigned to another unidentified compound present in the reaction bulk in very small quantities. In the 1H–1H COSY spectra, these additional signals are more clearly observed, and since they are all correlated with each other, it can be inferred that all of them belong to a single compound (see Figure 3).
The powder diffraction pattern of the starting reactive N-carbamoyl-L-proline (I) is shown in Figure 4a. It was indexed in an P212121 orthorhombic cell, with cell parameters a=6.4704(1) Ǻ, b=9.7644(1) Ǻ, and c=12.5135(2) Ǻ (refined values) [DICVOL04 (Boultif and Louër, Reference Boultif and Louër2004), M (20)=99.7 (de Wolff, Reference de Wolff1968), and F (20)=373.7 (Smith and Snyder, Reference Smith and Snyder1979)]. The cell parameters match those reported by Seijas et al. (Reference Seijas, Delgado, Mora, Bahsas and Briceño2007) from a single crystal study, and therefore a Rietveld refinement of the diffraction pattern was carried out using the structure reported by those authors; also, no additional phases were observed in it. The diffraction pattern of the reaction product shown in Figure 4b was indexed in an orthorhombic cell, with cell parameters a=11.376 53(3) Ǻ, b=8.001 12(2) Ǻ, and c=7.139 16(2) Ǻ (refined values) [DICVOL04 (Boultif and Louër, Reference Boultif and Louër2004), M (20)=1267.6 (de Wolff, Reference de Wolff1968), and F (20)=3877.7 (Smith and Snyder, Reference Smith and Snyder1979)]. The diffraction pattern showed eight additional peaks of very low intensity belonging to the unidentified compound also observed in the NMR experiments. Neither the NMR nor the diffraction experiment allowed the identification of the spurious phase, which might possibly be the product of the second reaction proposed in scheme 1. Evaluation of the systematic absent reflections of the major phase yielded space group P212121 (No. 19). The structural solution was carried out using the simulated annealing program DASH (David et al., Reference David, Shankland, van de Streek, Pidcock, Motherwell and Cole2006).

Figure 4. (Color online) Final Rietveld plot for (a) N-carbamoyl-L-proline (i) and (b) hydantoin-L-proline (IIa).
The starting molecule for the simulated annealing was obtained using the ab initio RHF/6-311G orbital optimization calculations implemented in GAUSSIAN03 (Frisch et al., Reference Frisch, Trucks, Schlegel, Scuseria, Robb, Cheeseman, Montgomery, Vreven, Kudin, Burant, Millam, Iyengar, Tomasi, Barone, Mennucci, Cossi, Scalmani, Rega, Petersson, Nakatsuji, Hada, Ehara, Toyota, Fukuda, Hasegawa, Ishida, Nakajima, Honda, Kitao, Nakai, Klene, Knox, Hratchian, Cross, Bakken, Adamo, Jaramillo, Gomperts, Stratmann, Yazyev, Austin, Cammi, Pomelli, Ochterski, Ayala, Morokuma, Voth, Salvador, Dannenberg, Zakrzewski, Dapprich, Daniels, Strain, Farkas, Malick, Rabuck, Raghavachari, Foresman, Ortiz, Cui, Baboul, Clifford, Cioslowski, Stefanov, Liu, Liashenko, Piskorz, Komaromi, Martin, Fox, Keith, Al-Laham, Peng, Nanayakkara, Challacombe, Gill, Johnson, Chen, Wong, Gonzalez and Pople2003). Cycles of ten million moves were performed and the program afforded a solution χ 2 of 16.4.
The model was refined against the diffraction pattern using the Rietvelt method (Rietveld, Reference Rietveld1969) implemented in the program GSAS (Larson and Von Dreele, Reference Larson and Von Dreele2004). Data in the range from 5° to 41.973° 2θ comprising 310 reflections were modeled with a pseudo-Voigt peak shape function (Thompson et al., Reference Thompson, Cox and Hastings1987), which includes the axial divergence correction at low angle (Finger et al., Reference Finger, Cox and Jephcoat1994). The background was described by the automatic linear interpolation of 20 points throughout the diffraction pattern. Restraints were applied to bond distance and bond angles with weights being 0.001–0.005 Ǻ and 1°, respectively. The restrained values for bond distances and angles were taken from the structure obtained by the ab initio RHF/6-311G calculations. The thermal motion was refined as follows: (a) an overall isotropic temperature factor for all the nonhydrogen atoms and (b) temperature factors for hydrogen atoms taken as 1.2 times the value of the nonhydrogen atoms. In the final Rietveld cycles the eight diffraction peaks corresponding to traces of the secondary unindexed phase were excluded from the pattern, improving the figures of merit by 3%. Details of the diffraction experiment, structural solution, and Rietveld refinement are summarized in Table I. Experimental, calculated, and differences plot for the final refinement cycle for compound (I) and (IIa) are shown in Figure 4. Table II shows
TABLE I. Crystallographic data and experimental details.

a selection of bond distances and angles for compound (IIa).
The analysis of the hydrogen bond network of N-carbamoyl-L-proline (I) (Seijas et al., Reference Seijas, Delgado, Mora, Bahsas and Briceño2007) shows hydrogen bonds, two of the type N---H⋯O and one O---H⋯O. These hydrogen bonds lock both the ureide and the acidic groups in an orientation that makes the NH2 group “anti” to the carboxylic acid group and at a distance of 4.310(3) Ǻ from its carbonylic carbon; therefore, any nucleofilic attack from the lone pair of the NH2 group to this carbon is geometrically hindered in the solid state. However, once the compound reaches it melting point, the free rotation of the ureide and the carboxylic acid groups is allowed and, therefore, a nucleophilic intramolecular substitution reaction might take place through the route shown in Figure 5 (Perreux et al., Reference Perreux, Loupy and Volatron2002).
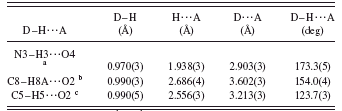
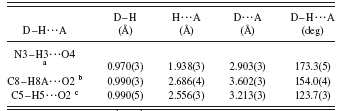
Once the reaction has taken place and the melt of the product solidifies, the molecular rearrangement caused by the reaction is so subtle that as a result the product (IIa) crystallizes in the same space group as the reactact (I), with more or less similar molecular packing but undergoing a cell contraction of 150 Å3 from the lost of four water molecules, one from each carbamoyl converted into the corresponding hydantoin in the unit cell. Figure 6 shows the molecular structure of the hydantoine-L-proline (IIa) with the atom labeling scheme. Bond distances in the same hydantoin [structural report without hydrogen atoms (Arte et al., Reference Arte, Tinant, Declercq, Germain and van Meerssche1980)] and other hydantoin rings (Yu et al., Reference Yu, Schwalbe and Watkin2004; Rizzi et al., Reference Rizzi, Schnur, Hutson, Kraus and Kelbaugh1989; Gauthier et al., Reference Gauthier, Yokum, Morales, McLaughlin, Liu and Fronczek1997; Delgado et al., Reference Delgado, Mora, Uzcátegui, Bahsas and Briceño2007) show similar asymmetry patterns as the ones reported here, with bond distances N1–C2 and N3–C4 being shorter by 0.057 Ǻ than N3–C2 bond distance. The asymmetry parameters of Griffin et al. (Reference Griffin, Duax and Weeks1984) evaluated in the pirrolidin ring show an envelope conformation, with C1 being the flap of the envelope [ΔCsmax=+47.1(3)°, ΔCsmin=+1.9(4)°, ΔC2max=+62.0(3)°, ΔC2min=16.5(3)°, ΔCs(C1)=1.9(4)°, and ΔCs(C4-N3)=1.9(4)°]. The crystal packing is stabilized by a hydrogen bond of the type N–H⋯O and two of the type C–H⋯O. The geometry of these hydrogen bonds is summarized in Table III. Pairs of molecules related by 21 screw axes interact by hydrogen bond N3–H3⋯O4 [1−x,−½+y,½−z], forming supramolecular chain structure described by the graph set C(4) (Etter, Reference Etter1990) (Figure 7). The repetition of these ribbons along a by means of another 21 screw axis produces sheets of molecules that pile up along the c direction. These sheets are linked by additional nonconventional hydrogen bonds of the
TABLE II. Selected bond distances (Å) and bond angles (deg) for hydantoin-L-proline (IIa).


Figure 5. Mechanism for the intramolecular nucleophilic attack from the lone pair of the NH2 group to the carboxylic acid group.

Figure 6. (Color online) Asymmetric unit and atom labeling scheme for the hydantoin-L-proline (IIa).
TABLE III. Hydrogen bonds in hydantoin-L-proline (IIa).
1 Symmetry code: 1−x,−½+y,½−z.
2 Symmetry codes: ½−x,−y,−½−z.
3 Symmetry codes: ½−x,−y,½+z.

Figure 7. (Color online) Partial packing view along c of hydantoin-L-proline (IIa). Broken lines indicate the hydrogen bonds. H atoms not involved in hydrogen bonding have been omitted for clarity. [Symmetry code: (i) 1−x,−½+y,½−z.]

Figure 8. (Color online) Packing view along (b) of hydantoin-L-proline (IIa). Broken lines indicate the hydrogen bonds. H atoms not involved in hydrogen bonding have been omitted for clarity. [Symmetry codes: (ii) ½−x,−y,−½−z; (iii) ½−x,−y,½+z.]
type C–H⋯O, C8–H8A⋯O2 [½−x,−y,−½−z], and C5–H5⋯O2 [½−x,−y,½+z], forming chains of fused supramolecular rings described by the graph set C21(6)R22 as shown in Figure 8.
IV. CONCLUSION
Conversion of N-carbamoyl-L-proline to hydantoin-L-proline was studied by means of X-ray powder diffraction experiments. Solvent-free in the solid state hydantoin-L-proline was obtained from direct heating of N-carbamoyl-L-proline via a nucleophilic intramolecular substitution reaction at 470 K. For this compound, a supramolecular chain structure is formed with a crystal packing directed by hydrogen bonds which displays a layered structure pile up along the c direction.
ACKNOWLEDGMENTS
We thank beamline ID31, ESRF for providing synchrotron radiation beam time, the CDCHT-ULA (Grant Nos. C-1618-08-08-AA and C-1614-08-08-B), and FONACIT (Grant No. LAB-97000821).