Introduction
Dilated cardiomyopathy (OMIM#115200) is a serious heart disorder and a leading cause of morbidity and mortality among young people worldwide.Reference Hershberger, Hedges and Morales 1 , Reference McNally and Mestroni 2 It is characterised by cardiac dilation and systolic dysfunction, and, in particular, left ventricular enlargement or dilatation.Reference Fan, Huang and Jin 3 – Reference Weintraub, Semsarian and Macdonald 5 Dilated cardiomyopathy is the major precipitating factor resulting in heart failure and sudden death.Reference Bezzina, Lahrouchi and Priori 6 The prevalence of dilated cardiomyopathy is more than 1 in 2500 individuals, and 30–50% of dilated cardiomyopathy cases are relatives.Reference Perez-Serra, Toro and Sarquella-Brugada 7
To date, mutations in more than 50 genes encoding the sarcomere, the Z-disk, the cytoskeleton, the mitochondria, RNA-binding proteins, the sarcoplasmic reticulum, and the nuclear envelope have been identified in dilated cardiomyopathy patients,Reference Hershberger, Hedges and Morales 1 , Reference Fan, Huang and Jin 3 such as Titin, Myosin Binding Protein C, Myosin Heavy Chain 7, and so on.Reference Haas, Frese and Peil 8 However, severe dilated cardiomyopathy caused by more than one gene is rare. Only two published papers report this phenomena.Reference Roncarati, Viviani Anselmi and Krawitz 9 , Reference Millat, Bouvagnet and Chevalier 10 Research on dilated cardiomyopathy patients carrying more than one diseasing-causing mutation has the potential to expand our understanding about the genes and the molecular mechanisms involved in dilated cardiomyopathy.
In this study, we have enrolled a family diagnosed with dilated cardiomyopathy from the central south of China. The proband and his parents all showed dilated cardiomyopathy phenotypes, but the condition of the proband was more severe. Whole-exome sequencing was applied to identify the genetic lesion of this family.
Materials and Methods
Subjects
The study protocol was approved by the Review Board of the Second Xiangya Hospital of the Central South University in China, and the study participants gave informed consent. A total of 12 members of the family, with six affected individuals, were enrolled in this study. Blood was obtained from the affected probands and family members. Subjects’ medical records and echocardiography were reviewed.
Whole-exome sequencing
Genomic DNA was extracted from peripheral blood lymphocytes of all the family members. Genomic DNA was prepared using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, California, United States of America) as we have described previously.Reference Xiang, Fan and Huang 11
The molecular analysis of the proband was performed by whole-exome sequencing. The main part of whole-exome sequencing was provided by the Novogene Bioinformatics Institute (Beijing, China). The exomes were captured using Agilent SureSelect Human All Exon V6 kits (Agilent Genomics, Santa Clara, California, United States of America), and the platform of high-throughput sequencing was performed in Illumina HiSeq X-10 (Illumina, San Diego, California, United States of America). The basic bioinformatics analyses including Reads, Mapping, Variant detection, Filtering, and Annotation were also endowed by Novogene Bioinformatics Institute as we have described.Reference Liu, Fan and Zhang 4
Mutation validation and co-segregation analysis
All the filtered mutations and co-segregation analysis of the dilated cardiomyopathy family members were validated by Sanger sequencing. The primer pairs were designed by Primer 5 – the sequences of primers will be provided upon request – and sequences of the polymerase chain reaction products were determined using the ABI 3100 Genetic Analyzer (ABI, Foster City, California, United States of America).
Results
Clinic data
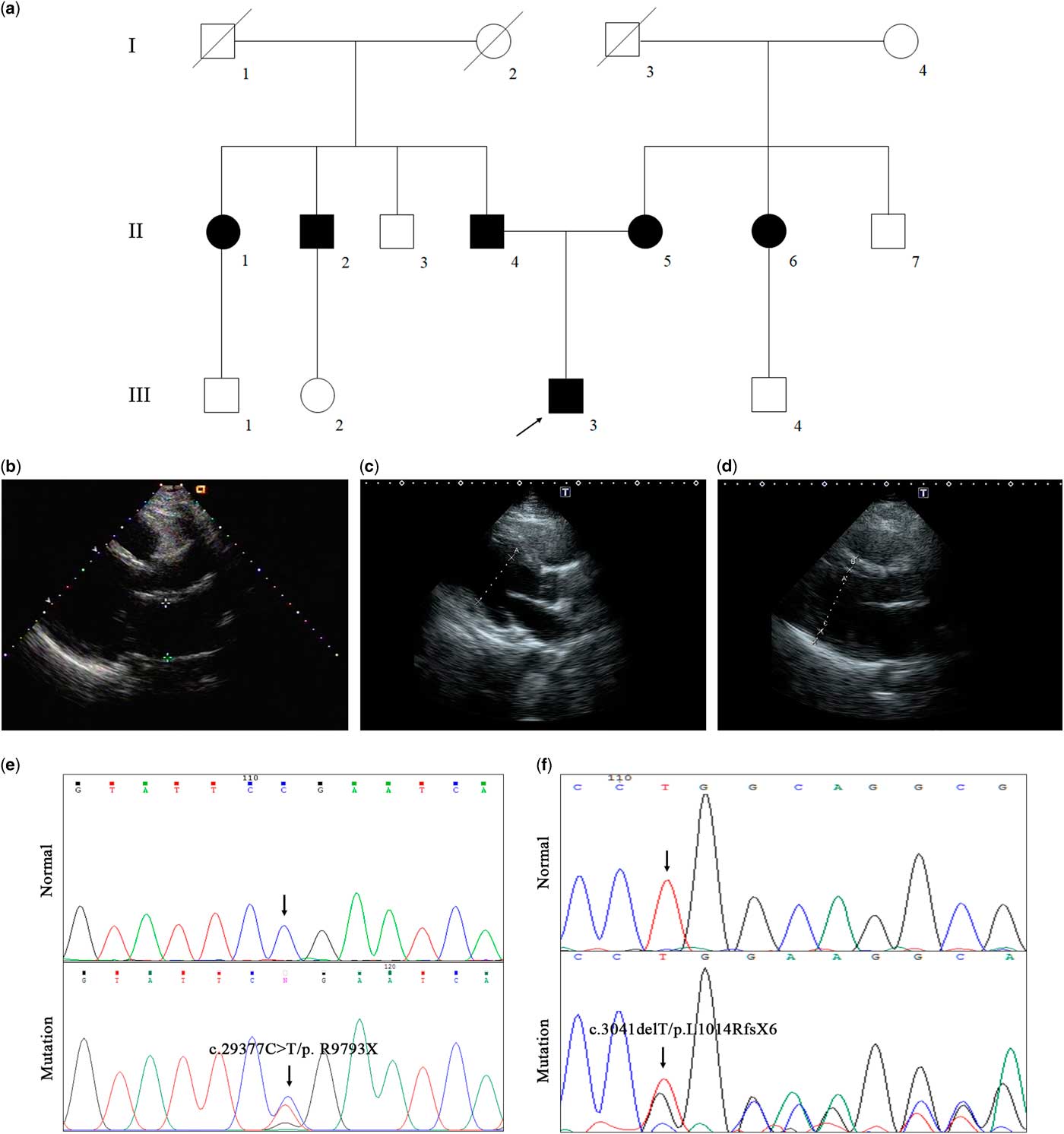
We have enrolled a Chinese family with dilated cardiomyopathy and syncope (Fig 1a, Table 1). The proband (III-3), a 15-year-old boy from Jiangxi province of Central–South China, experienced syncope during stair-climbing. Echocardiography showed an obvious enlargement of the left ventricle (70 mm) and left atrium (57 mm) (Fig 1b, Table 1). Case history investigation revealed that both his mother and father suffered from heart enlargement but without syncope. Echocardiography confirmed the dilated cardiomyopathy phenotypes of his parents (Fig 1c and d). In addition, two of the proband’s aunts and one uncle also suffered from heart enlargement (Table 1).

Figure 1 The clinical and genetic data of the dilated cardiomyopathy family. ( a ) Pedigree of the family. Family members are identified by generations and numbers. Squares indicate male family members; circles, female members; closed symbols, affected members; open symbols, unaffected members; and the arrow, the proband. The B ultrasonic data of the proband ( b ), his mother ( c ), and his father ( d ). ( e ) Sanger DNA sequencing chromatogram demonstrates the heterozygosity for a Titin mutation (c.29377 C>T/ p. R9793X). ( f ) Sanger DNA sequencing chromatogram demonstrates the heterozygosity for a Myosin Binding Protein C mutation (c.3041delT/p.L1014RfsX6).
Table 1 The clinical data of the family.

AO=aorta dimension; FS=fractional shortening; IVS=left ventricular septum thickness; LA=left atrial diameter; LV=left ventricle; LVPW=left ventricular posterior wall; PA=pulmonary artery inner diameter; RA=right atrial diameter; RV, right ventricle
Genetic analysis
Whole-exome sequencing yielded 9.54 Gb data with 99.7% coverage of the target region and 99.1% of the target covered over 10×. In total, 81,042 variants were present in the proband. After 1000 G and dbSNP132 database filtering, only 573 variants were retained. The remaining variants were further filtered by cardiomyopathy-related genes (Supplementary Table S1), and a set of 12 variants in seven genes were detected. All the 12 variants were predicted by three bioinformatic programmes (MutationTaster, SIFT, and Polyphen-2) (Table 2).
Table 2 The gene list of Sanger sequencing validation and co-segregation analysis.

AB=alternative base identified; B=benign; CHR=chromosome; D=disease-causing; N=polymorphism; P=probably damaging; POS=position; RB=reference sequence base; T=tolerated Blue words represent healthy family members
We then designed the related primers to perform Sanger sequencing and co-segregation analysis. Surprisingly, the proband’s father, one uncle, and one aunt all carried novel mutations (c.29377 C>T/ p. R9793X) of Titin (Fig 1e), and both the proband’s mother and one aunt carried a novel mutation (c.3041delT/p.L1014RfsX6) of Myosin Binding Protein C (Fig 1f). Both mutations, Myosin Binding Protein C: p.L1014RfsX6 and Titin: p. R9793X, were identified in the proband (Fig 1e and f). No other healthy family members carried either mutation.
The deletion mutation, c.3041delT/p.L1014RfsX6, presents a premature stop codon at position 1020 in exon 28 of the Myosin Binding Protein C gene, which leads to truncation of the protein. The nonsense mutation, c.29377 C>T/ p. R9793X, of Titin is located in the highly evolutionarily conserved domain, resulting in truncation of the protein. Neither novel mutation was found in our 200 local control cohorts.Reference Fan, Huang and Jin 3
Discussion
In this study, a family from Central–South China diagnosed with dilated cardiomyopathy was enrolled to investigate the genetic lesion of the affected individuals. By whole-exome sequencing in combination with a cardiomyopathy-related gene filtering strategy, we detected two novel mutations – Myosin Binding Protein C: p.L1014RfsX6 and Titin: p.R9793X – in the proband. Co-segregation analysis further confirmed that both mutations were disease-causing variants and would lead to dilated cardiomyopathy in the affected individuals. Our study reported a unique dilated cardiomyopathy case caused by both novel heterozygous Myosin Binding Protein C and Titin mutations. Our study will provide more information about the disease-causing genes associated with dilated cardiomyopathy.Reference McNally, Golbus and Puckelwartz 12
Previous studies demonstrated more than nine hundred mutations of Myosin Binding Protein C in cardiomyopathy patients.Reference van Velzen, Schinkel and Oldenburg 13 It is estimated that ~35% of all cardiomyopathy cases in Western countries are due to Myosin Binding Protein C mutations.Reference Liu, Jiang and Piao 14 Myosin Binding Protein C can bind F-actin and native thin filaments and modify the activity of actin-activated myosin ATPase.Reference Gresham and Stelzer 15 , Reference Kumar, Govindan and Zhang 16 It may modulate muscle contraction or play a structural role in the sarcomere.Reference Ito, Patel and Gorham 17 This novel mutation, c.3041delT/p.L1014RfsX6, of Myosin Binding Protein C locates in the Ig-Titin-like domain and results in the loss of fibronectin type 3 domain. These two domains can bind to Titin protein and stabilise the structure of the sarcomere.Reference Kuster, Govindan, Springer, Martin, Finley and Sadayappan 18 The deletion mutation, c.3041delT/p.L1014RfsX6, may disturb the structure of the sarcomere by affecting these two domains of Myosin Binding Protein C and lead to dilated cardiomyopathy.
Titin has multiple critical roles in all striated muscle cells.Reference Chauveau, Rowell and Ferreiro 19 It acts as an architectural protein and provides particular attachment to a plethora of essential proteins such as actin, CARP, and Obscurin.Reference Liu, Fan and Zhang 4 Titin consists of four structurally and functionally distinct regions: the amino-terminal Z-line, the I-band and A-band regions, and the carboxy-terminal M-line extremity. The novel mutation (p.R9793X) of Titin causes a truncation of the protein and affects the I-band region, A-band region, and M-line region. The function of Titin may be completely lost and eventually result in dilated cardiomyopathy.Reference Liu, Fan and Zhang 4 , Reference Chauveau, Rowell and Ferreiro 19
In our study, both mutations of Myosin Binding Protein C and Titin can result in truncation of the protein. Previous analysis indicated that the sarcomere of cardiomyocytes in the proband may be disrupted more severely than that in the affected individuals with either isolated mutation. Consistently, the phenotypes of the proband were more severe than those of other individuals: only the proband had a family history of syncope; the proband was the youngest one who suffered from dilated cardiomyopathy and his heart was significantly enlarged. These observations may further prove that the proband’s severe dilated cardiomyopathy was caused by both mutations of Myosin Binding Protein C and Titin.
In conclusion, by using whole-exome sequencing, we have identified two novel heterozygous mutations – Myosin Binding Protein C: p.L1014RfsX6 and Titin: p.R9793X – in a severe dilated cardiomyopathy patient. Co-segregation analysis further confirmed that the mutation (c.3041delT/p.L1014RfsX6) of Myosin Binding Protein C came from his mother and the mutation (c.29377 C>T/ p. R9793X) of Titin came from his father. Both mutations were first reported in dilated cardiomyopathy patients. Our study not only provides a unique case to understand the genes and the molecular mechanisms involved in the phenotype of dilated cardiomyopathy but also expands the spectrum of Myosin Binding Protein C and Titin mutations and may contribute to the genetic diagnosis and counselling of dilated cardiomyopathy patients.
Acknowledgements
The authors thank all subjects for participating in this study.
Financial Support
This study was supported by the National Natural Science Foundation of China (81470445).
Conflicts of Interest
None.
Ethical Standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines of China on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008, and has been approved by the Review Board of the Second Xiangya Hospital of Central South University.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1047951118001403