Introduction
Chinese jiaotou (Allium chinense), also known as Rakkyo in Japan, is a tetraploid (2n = 4X = 32) perennial plant that belongs to the Liliaceae. The species is an economically important crop, and it not only is consumed as a vegetable (Xu et al., Reference Xu, Um, Kim, Lu, Guo, Liu, Bah and Mao2008) but also is used in the treatment of many diseases (Mann and Stearn, Reference Mann and Stearn1960). Because of its nutritional and medicinal properties, Chinese jiaotou is widely cultivated in East Asian countries, especially China and Japan, and the crop has been cultivated in China for more than 3000 years (Mann and Stearn, Reference Mann and Stearn1960).
In China, there are large numbers of local varieties of Chinese jiaotou, which exhibit variation in many morphological features, including bulb shape. However, few molecular markers have been described, which hinders the accurate evaluation of the genetic diversity of these varieties. Although Chinese jiaotou can still be investigated using universal molecular markers, such as random amplified polymorphism DNA (RAPD), amplified fragment length polymorphisms (AFLPs), and sequence-related amplified polymorphism (SRAP), these markers possess common shortcomings, which include poor repeatability and dominance. In contrast, simple sequence repeat markers (SSRs) possess several advantages, including reproducibility, multiple alleles, codominance and simple analysis (Liu et al., Reference Liu, Zeng, Zhu, Chen, Tang, Mei and Tang2015a). Therefore, SSR markers have been used extensively for genetic and breeding studies in many crops (Liu et al., Reference Liu, Mao, Zhang, Xu and Xing2009, Reference Liu, Zhu, Tang and Tang2014a; Li et al., Reference Li, Ma, Chen, Liu, Shen, Tu, Xing and Fu2012; Zhu et al., Reference Zhu, Zheng, Dai, Tang and Liu2016).
One of the main reasons that few SSRs have been developed for Chinese jiaotou is the crop's paucity of genomic and genic sequences, which is likely a result of the crop's large genome (approximately 16 Gb; Ohri et al., Reference Ohri, Fritsch and Hanetl1998), and despite the recent emergence of next-generation sequencing (NGS) technology, which provides a powerful and cost-efficient tool for sequence determination, the crop's genome size has still hindered genomic characterization. In contrast, transcriptome analysis by NGS is also rapid and inexpensive but is unlimited by genomic size or complexity. Accordingly, the technique has been widely used as a primary tool in a variety of research areas, including gene discovery (Liu et al., Reference Liu, Zhu, Tang and Tang2014b, Reference Liu, Zhu, Tang and Tang2015b; Zheng et al., Reference Zheng, Tang, Zhu, Dai and Liu2016a), crop domestication pattern (Liu et al., Reference Liu, Zhu, Tang, Chen, Yu and Tang2013a, Reference Liu, Tang, Zhu, Tang and Zheng2014c), and expression profile analysis (Liu et al., Reference Liu, Zhu, Tang and Tang2015c; Yu et al., Reference Yu, Zeng, Yan, Liu, Sun, Zhu and Zhu2015; Mei et al., Reference Mei, Liu and Wang2016; Zeng et al., Reference Zeng, Shen, Chen, Yan, Liu, Xue and Yu2016). In addition, transcriptome analysis is also a powerful tool for the large-scale development of SSRs based on assembled expressed sequence tags (ESTs), and to date, transcriptome analysis has been used to develop large numbers of SSRs in many plant species (Liu et al., Reference Liu, Zhu, Fu, Tang, Yu, Chen, Luan, Wang and Tang2013b; Ding et al., Reference Ding, Jia, Luo, Zhang, Cong, Liu and Bai2015; Zhang et al., Reference Zhang, Li, Tao, Fang and Qi2015).
Recently, the transcriptome of Chinese jiaotou had been sequenced and de novo assembled using Illumina paired-end sequencing technology, resulting in 121,008 non-redundant ESTs (Genebank number: GFAL00000000; Zhu et al., Reference Zhu, Tang, Tan, Yu, Dai and Liu2017a). In the present study, based on these ESTs, SSR markers of Chinese jiaotou were developed. Thereafter, these developed SSRs were assessed for their cross-species transferability, and used to analyse the genetic relationships of Chinese jiaotou accessions.
Materials and methods
Plant materials and nucleic acid extraction
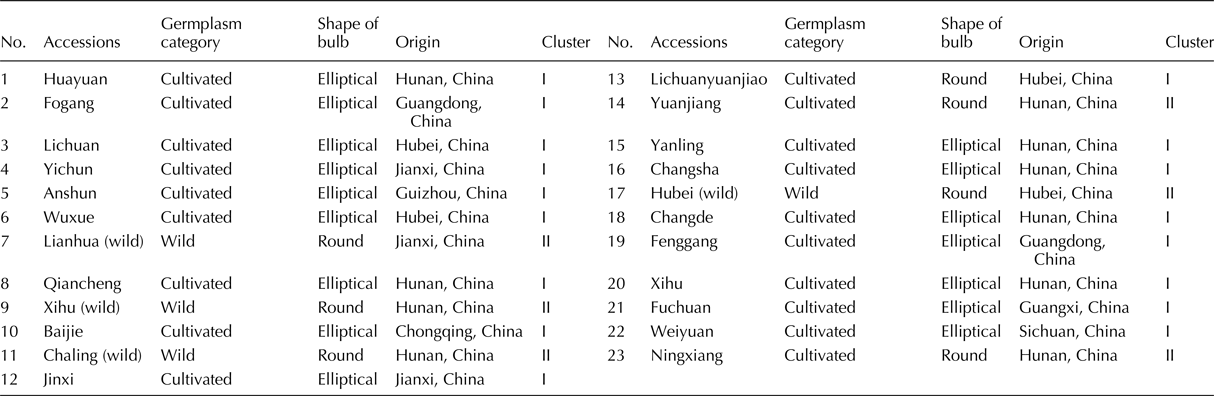
A local variety of Chinese jiaotou from Ningxiang (Changsha, China) and six related Allium crops (Chinese chive, shallot, garlic, leek, onion and Welsh onion) were planted in the experimental field of the Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences (Changsha, China), in September 2014. Four wild and 19 cultivated accessions from eight provinces of China (Table 1) were also grown in this experimental field, in September 2016. Fresh leaves of Chinese jiaotou, its six relatives and the 23 accessions were collected, and DNA was extracted using a Plant Genomic DNA Kit (TIANGEN, China), according to the manufacturer's protocol.
Table 1. Accessions used for diversity analysis
Identification of SSR loci and development of markers
The transcriptome sequences were downloaded from the Genebank database by the accession number of GFAL00000000. Putative SSRs were identified from the ESTs of Chinese jiaotou using MISA 1.0 (Thiel, Reference Thiel2003), with the default criteria: a minimum of seven repeats for dinucleotide motifs and a minimum of five repeats for tri-, tetra- and pentanucleotide motifs. Primers flanking the putative SSRs were then designed using Primer 3.0 software (Untergasser et al., Reference Untergasser, Nijveen, Rao, Bisseling, Geurts and Leunissen2007) with the following criteria: length, 17–23 bp; GC content, 40–60%; and estimated amplicon size, 100–300 bp. To determine the location of SSR in EST, the coding sequence (CDS) of each transcript was predicted by BLAST searching against NCBI non-redundant protein sequence and SwissProt protein databases, as well as by estscan program (Iseli et al., Reference Iseli, Jongeneel and Bucher1999). GO functional classification for the SSR-containing sequences were performed by the WEGO software (Ye et al., Reference Ye, Fang, Zheng, Zhang, Chen, Zhang, Wang, Li, Li, Bolund and Wang2006). The enrichment of GO functional categories was analysed using GOseq based on the Wallenius noncentral hypergeometric distribution (Young et al., Reference Young, Wakefield, Smyt and Oshlack2010). Q values were used to determine the P-value threshold in multiple tests and analysis, and GO categories with Q < 0.05 were considered significantly enriched.
Amplification of SSR-containing regions
The quality of the developed SSR primer pairs was assessed, and their cross-species transferability was characterized. A total of 100 SSR markers (CHM001–CHM100) were selected for amplification in Chinese jiaotou and six related Allium crops (Chinese chive, shallot, garlic, leek, onion and Welsh onion). In addition, these SSRs were used for PCR amplification in 23 accessions to evaluate their genetic diversity. PCR amplification was conducted in 10-μl reaction mixtures that contained 1 µl genomic DNA (~30 ng/μl), 0.8 µl dNTP mix (2.5 mM), 0.6 µl of the specific primer (10 pmol/μl), 1.0 µl 10× rTaq PCR buffer (Laifeng, China) and 0.2 µl rTaq polymerase (5 unit/μl; Laifeng). PCR reaction conditions followed previous studies (Liu et al., Reference Liu, Shao, Kovi and Xing2010a, Reference Liu, Zhang, Zhang and Xing2011a). The SSR assay was carried out according to the method described by Wu and Tanksley (Reference Wu and Tanksley1993).
Analysis of genetic diversity of 23 accessions
The SSR markers were used to assess the genetic relatedness of 23 accessions (Table 1). The allelic data were converted into a binary matrix, with the scores 1 and 0 denoting the presence or absence of a given allele, respectively. The data were analysed using the Numerical Taxonomy Multivariate Analysis System (NTSYS-pc) version 2.10 (Rohlf, Reference Rohlf2002) and genetic similarity (GS) coefficients were calculated from the coefficient for similarity using the software's SIMQUAL module. A dendrogram was then constructed from the resulting GS matrix, using the unweighted pair group method with arithmetic average (UPGMA) to determine genetic relationships among these accessions.
Results
Development and characterization of SSR markers
A total of 2157 SSR loci were identified from the 121,008 EST sequences of Chinese jiaotou, with a total length of 66.84 Mb, which suggested that the EST–SSRs occurred at a frequency of one SSR per 31 kb of EST sequence. Of these 2157 loci, 663 were located at the ends of EST sequences, which complicated the design of primer pairs for flanking regions. Ultimately, primer pairs were designed for 1494 of the SSR loci, and were named CHM0001 to CHM1494, respectively (Supplementary Table S1).
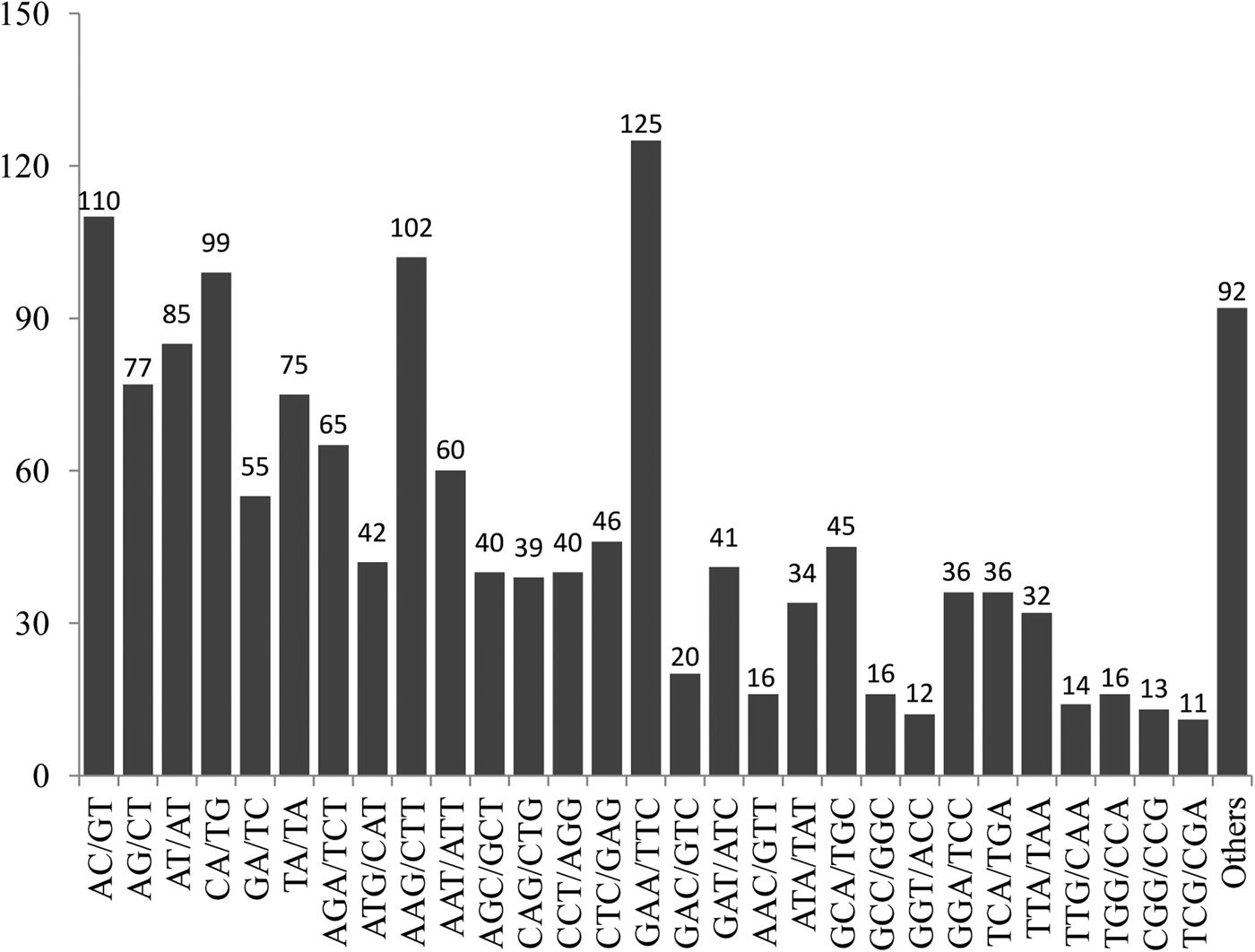
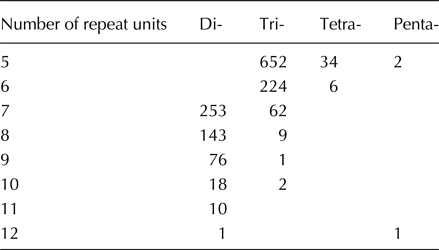
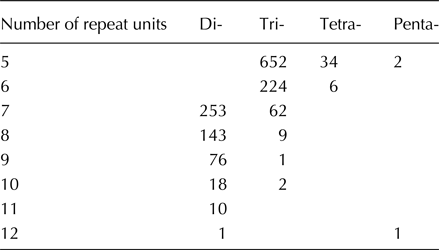
Among the 1494 markers, trinucleotide repeat motifs were the most abundant (950, 63.6%), followed by dinucleotide repeat motifs (501, 33.5%), and only 40 tetranucloetide, three pentanucleotide and zero hexanucleotide markers were identified (Table 2). Most of the SSRs (90.2%) ranged from 14 to 18 bp in length (Table 2). In addition, 69 motif sequence types were identified, including 6, 34, 26 and 3 di-, tri-, tetra- and pentanucleotide repeats, respectively. GAA/TTC trinucleotide repeats (125, 8.37%) were the most abundant, followed by AC/GT (110, 7.36%), AAG/CTT (102, 6.83%), CA/TG (99, 6.63%), AT/AT (85, 5.69%), AG/CT (77, 5.15%) and TA/TA (75, 5.02%) motifs, respectively (Fig. 1).
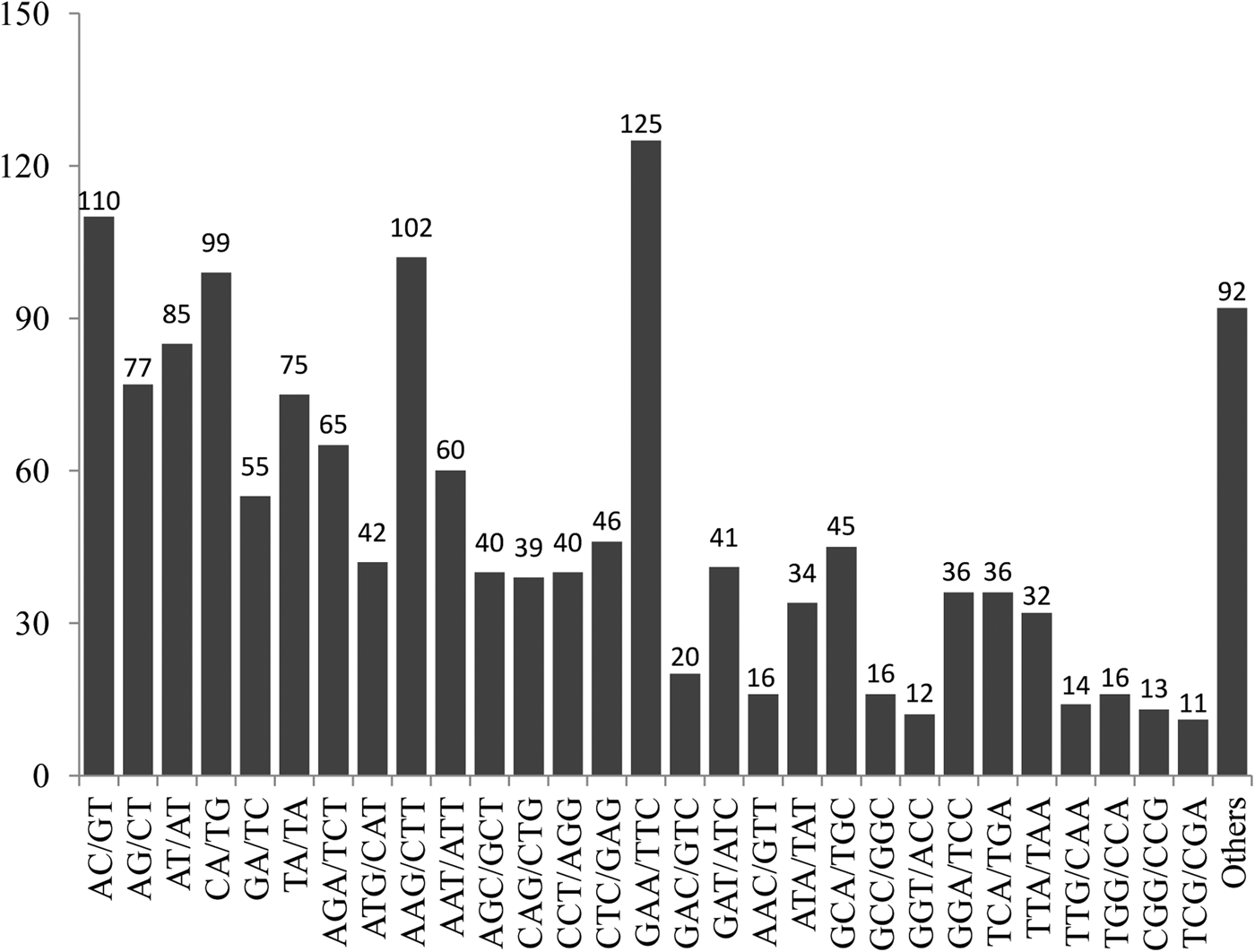
Fig. 1. Frequency distribution of EST–SSRs based on motif sequence types.
Table 2. Length distribution of EST–SSRs based on the number of repeat units
Annotated function of the SSR-containing ESTs
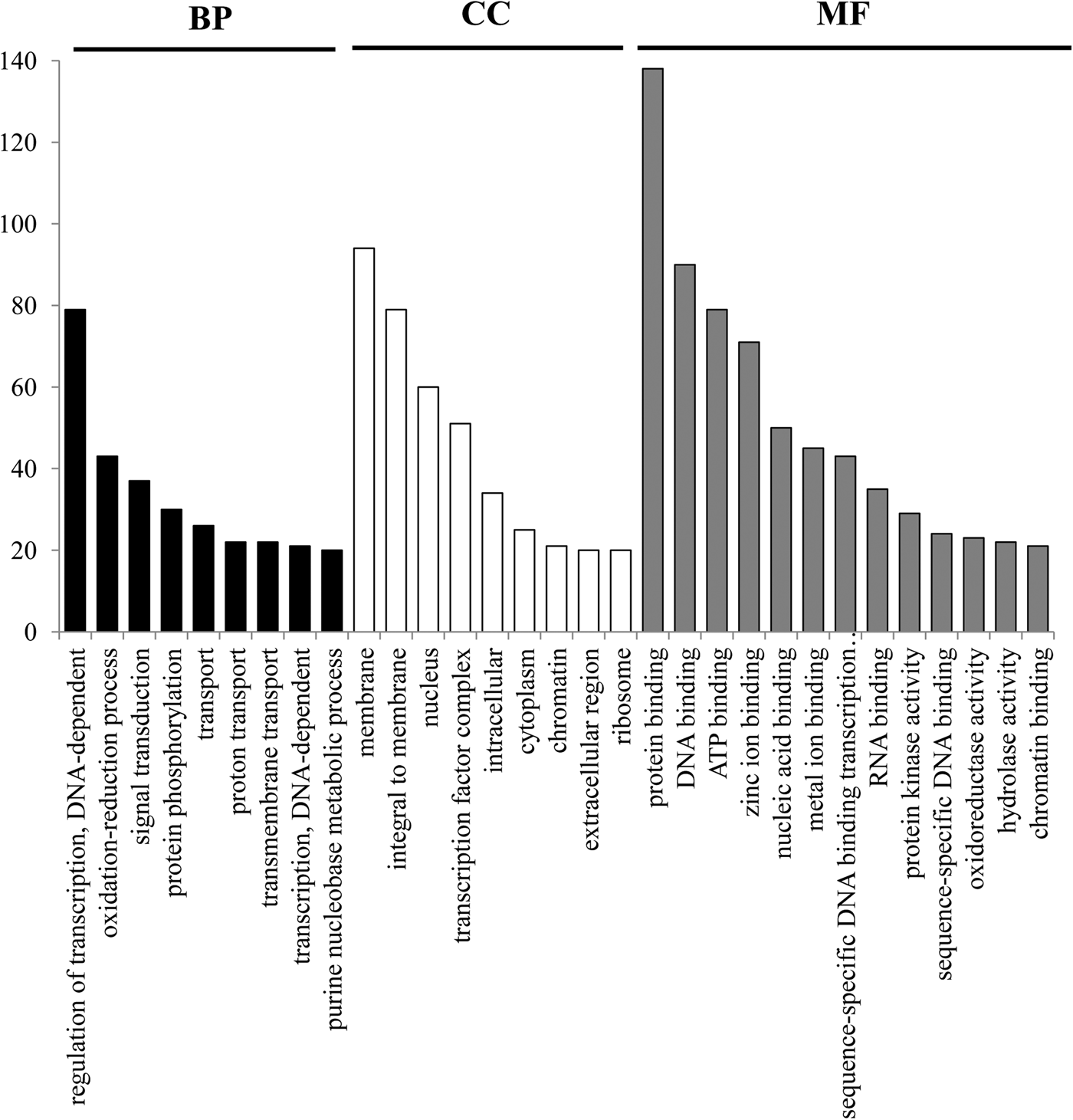
The EST sequences in which these 1494 SSRs were contained were used to investigate their function annotated. The result showed that 1494 SSRs fell into 1459 sequences, of which 624, 257 and 332 SSRs fell into the CDS, 5′ untranslated region (5′ UTR), and 3′ untranslated region (3′ UTR), respectively (Supplementary Table S1). However, there were 281 loci with an uncertain location in the ESTs, because the CDS of these ESTs cannot be determined. Among these 1459 SSR-containing ESTs, 906 were achieved functional annotation by Zhu et al. (Reference Zhu, Tang, Tan, Yu, Dai and Liu2017a, Supplementary Table S2), of which 93 were identified as transcription factor-encoding genes. The GO functional classification of these SSR-containing ESTs revealed that these sequences matched to known proteins of GO database with 3311 functional terms. Of these terms, there were 1206, 702 and 1403 were assigned to the biological process, cellular component and molecular function ontologies, respectively (Fig. 2). Interestingly, several categories involved in transcription regulation, oxidation–reduction, transport, etc. showed significant enrichment by these sequences (Q < 0.05).
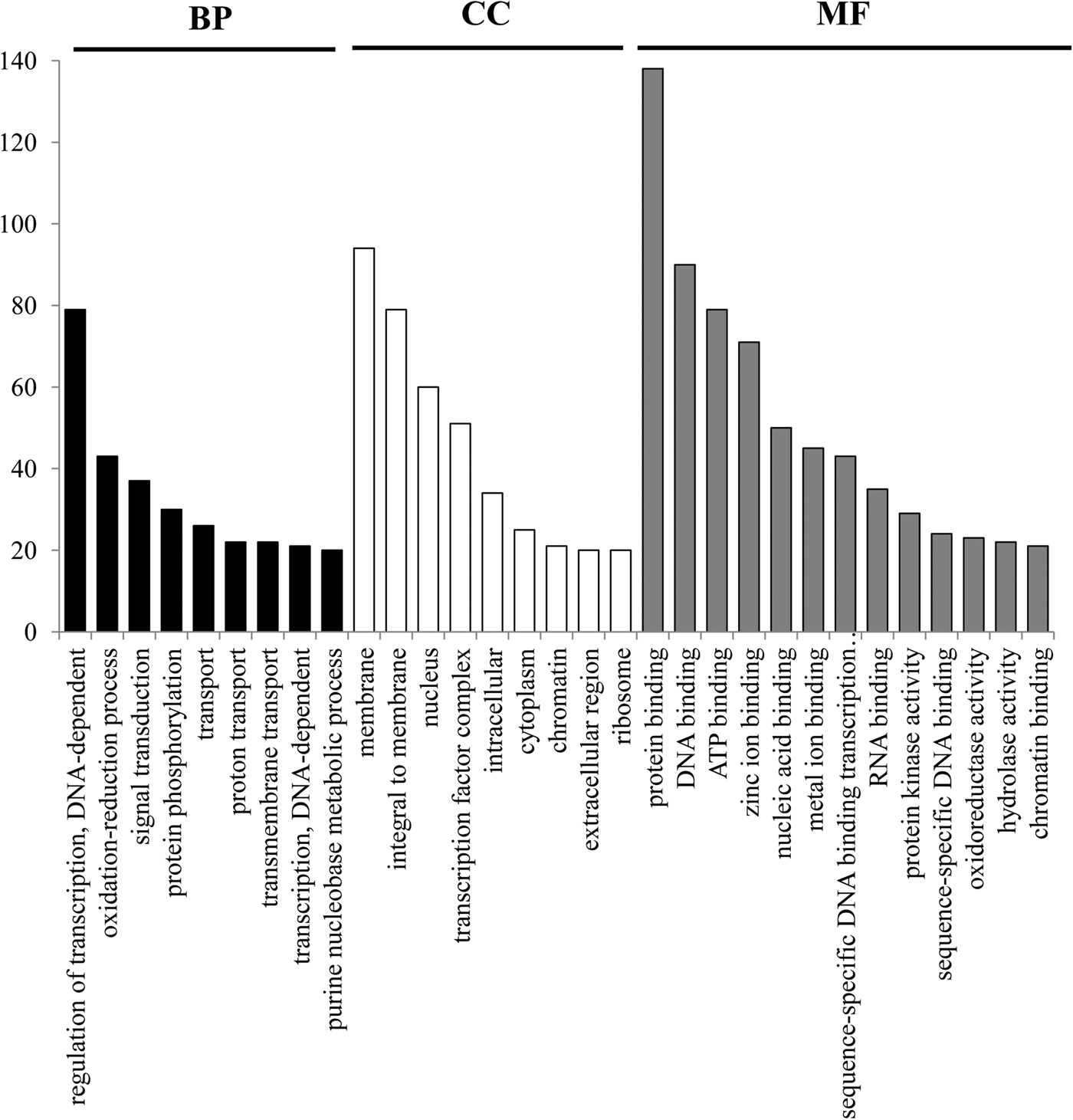
Fig. 2. Gene ontology classifications of SSR-containing ESTs. Only the functional category with more than 20 terms was showed in the figure, and the red, green, and blue histogram bar represented three main categories: biological process, cellular component and molecular function.
Evaluation of the SSR marker quality and transferability
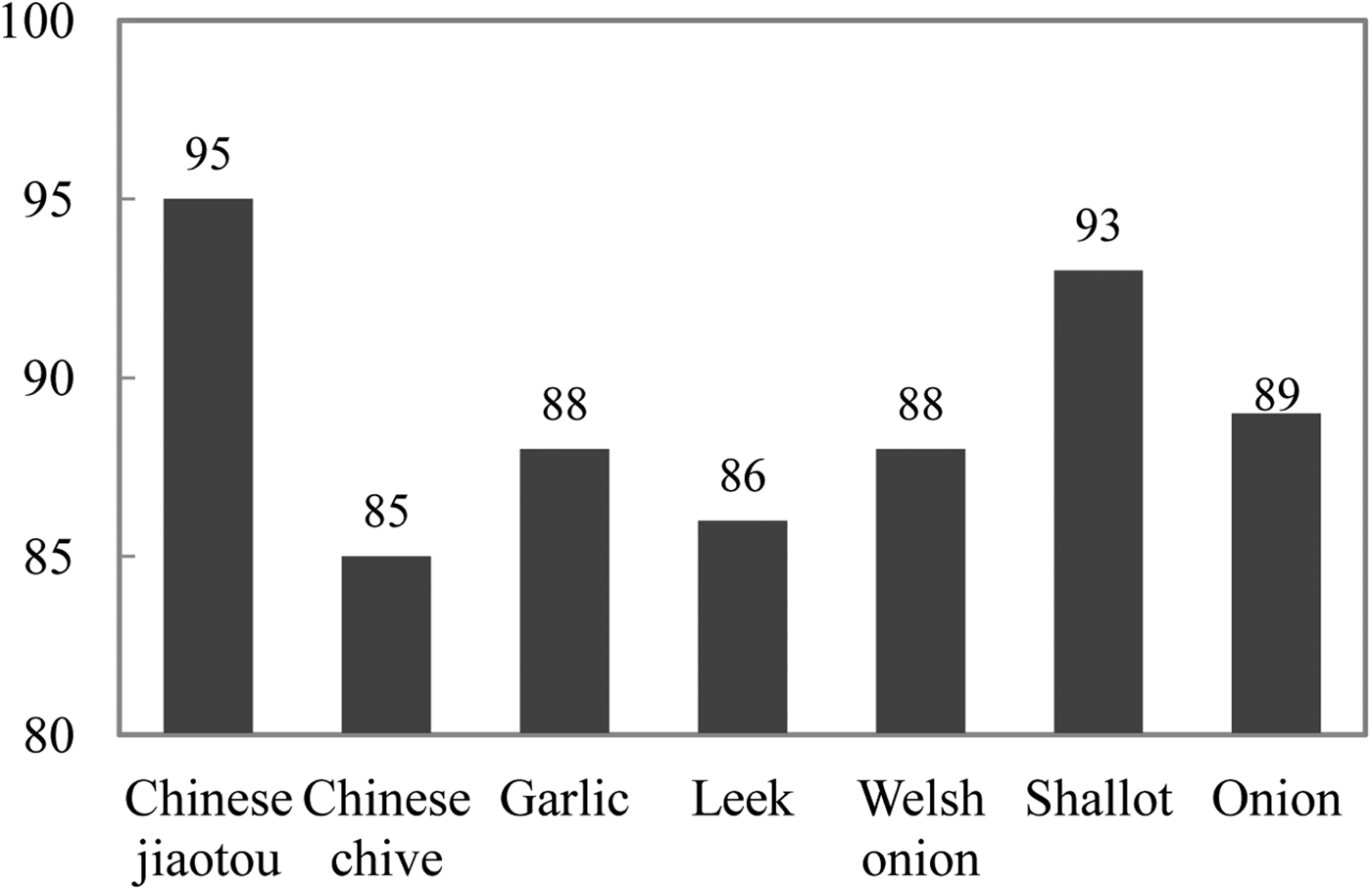
To assess the quality of the developed SSR markers, 100 SSRs (CHM001–CHM100) were randomly chosen to amplify using polymerase chain reaction (PCR). Finally, 95 of the 100 representative primer pairs could achieve successful amplification (Supplementary Table S2). In addition, the transferability of these 100 SSR markers to six other Allium species was analysed. Of these 100 SSRs, the primer pairs of 85 amplified fragments from Chinese chive, and 88 in garlic, 86 in leek, 88 in Welsh onion, 93 in shallot and 89 in onion achieved successful amplification, respectively (Fig. 3, Supplementary Table S3). Taken together, 69 of the Chinese jiaotou markers were transferable to all six other Allium species, and 97 markers amplified fragments from at least one of the six other Allium species. This suggested that the SSR markers possessed a good cross-species transferability.
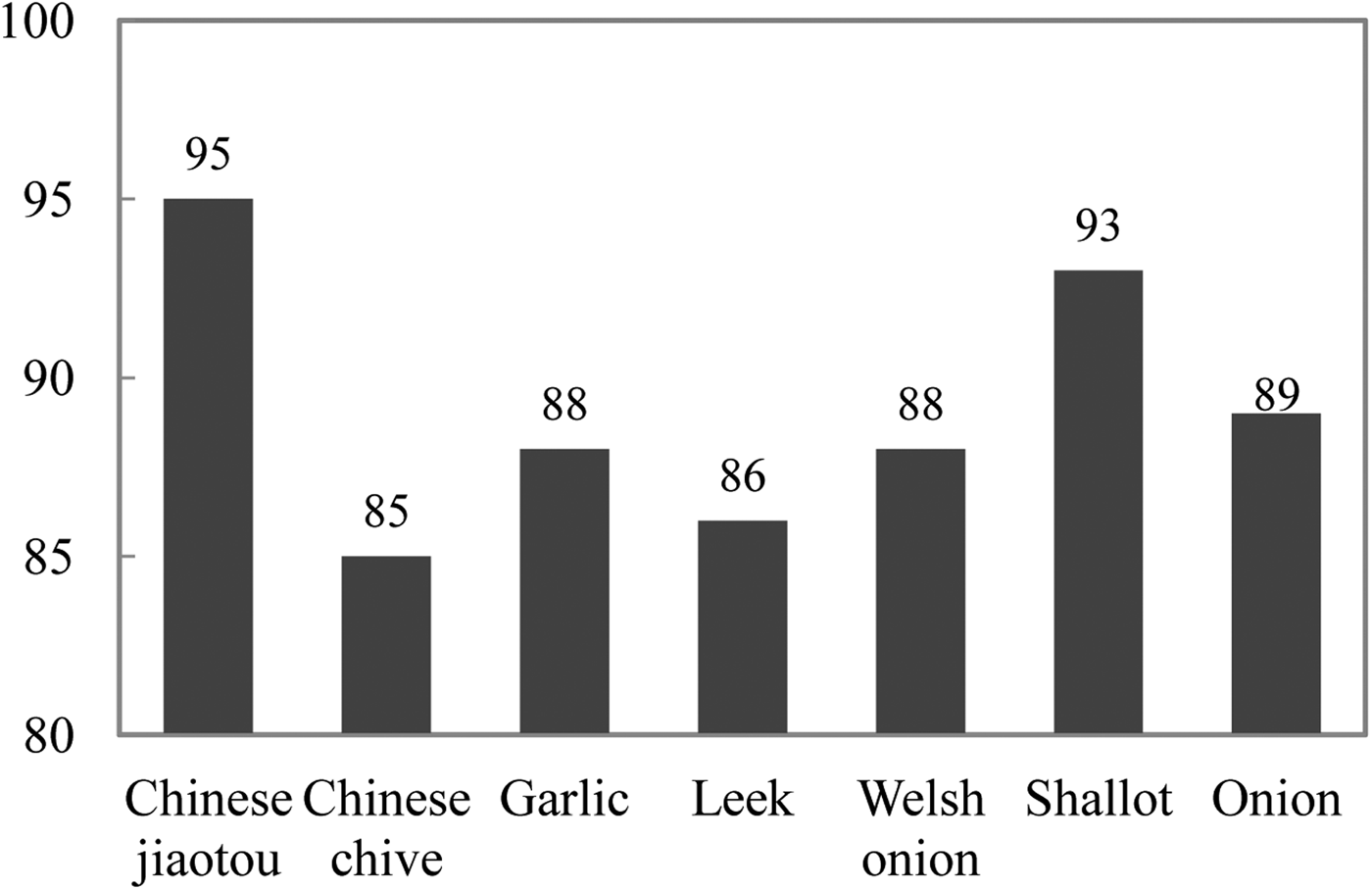
Fig. 3. Number of SSR markers successfully amplified in Chinese jiaotou and six related Allium crop species among 100 putative SSRs.
Genetic relatedness of 23 accessions
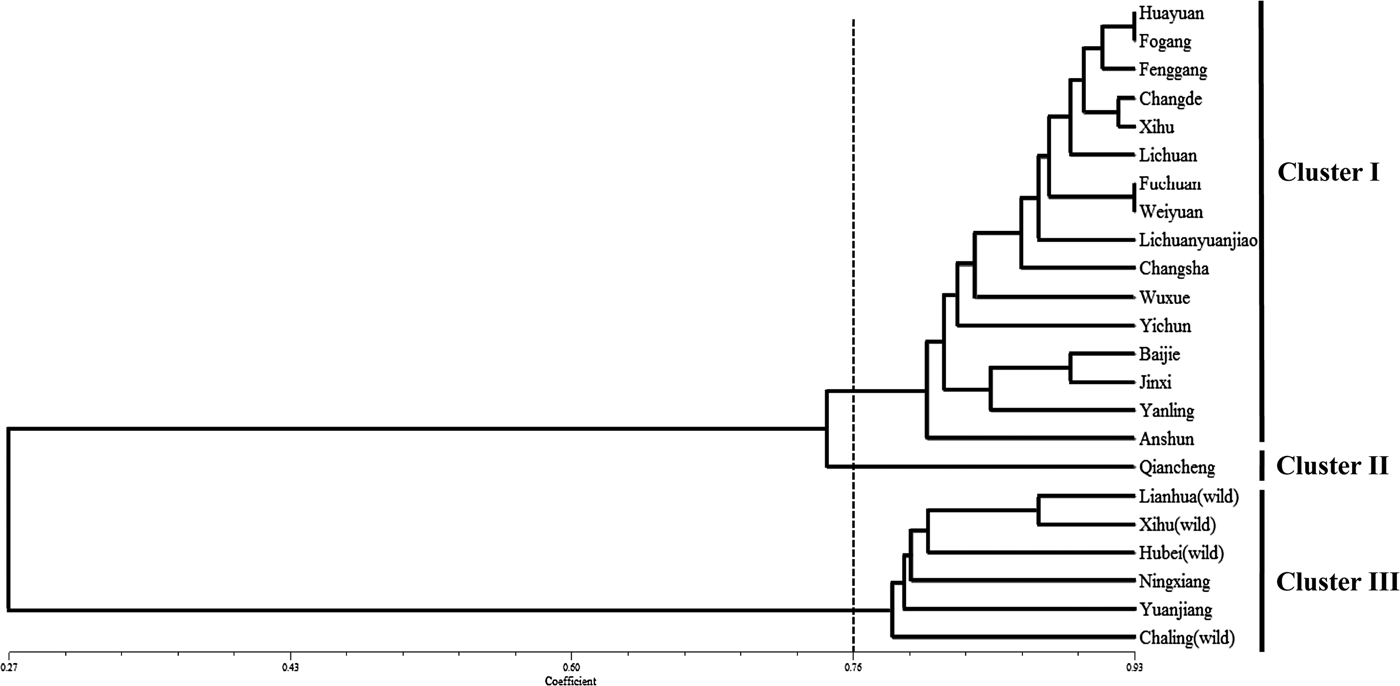
The genetic diversity and relatedness of 23 accessions (three wild and 19 local accessions) from eight provinces of China were analysed (Table 1). All four wild accessions and three of the 19 cultivated genotypes produce round bulbs, whereas the other 16 cultivated genotypes produce elliptical bulbs, respectively (Table 1). The 95 primer pairs that were suitable for amplification in Chinese jiaotou (Supplementary Table S3) were used to evaluate the genetic relationships among the 23 accessions, using similarity coefficients. The resulted showed that the similarity coefficients ranged from 0.27 to 0.93. Taking a similarity coefficient of 0.76 as the threshold, 23 accessions could be distinctly classified into three clusters (Fig. 4). The main group, cluster I, included 16 of the varieties and cluster III included six. Cluster II comprised only one variety, Qiancheng. Interestingly, 15 of the 16 varieties in cluster I produced elliptical bulbs, and all six accessions in cluster III produced round bulbs (Table 1). In addition, all four wild genotypes were assigned to cluster III, and two local varieties from northern Hunan were closely related to the four wild genotypes.
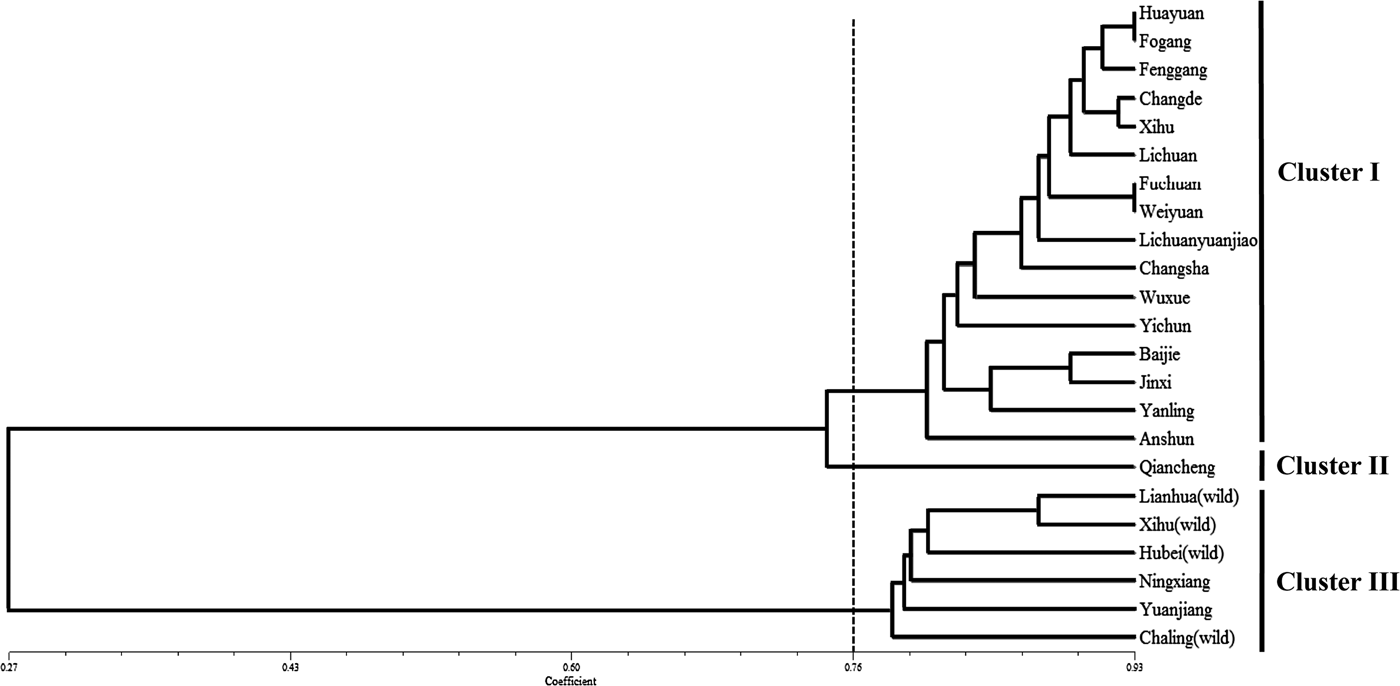
Fig. 4. Genetic relatedness of 23 accessions of Chinese jiaotou, based on the cluster analysis of 95 EST–SSR markers.
Discussion
Development of SSR markers in Chinese jiaotou
In contrast to RAPD, SRAP and AFLP, which produce non-specific amplicons, SSR markers possess a variety of advantages, including reproducibility, multiple alleles, codominance and simple analysis (Liu et al., Reference Liu, Zeng, Zhu, Chen, Tang, Mei and Tang2015a). Accordingly, SSR markers have been used as a primary tool for genetic mapping, identifying varieties with agronomic potential, characterizing and certifying plant materials, and crop-breeding (Liu et al., Reference Liu, Zhang, Xue, Xu, Li and Xing2010b, Reference Liu, Li, Zhang, Xu, Li and Xing2011b; Mao et al., Reference Mao, Liu, Xu, Li and Xing2011; Zhu et al., Reference Zhu, Liu, Dai, Wu, Zheng, Tang and Chen2017b). However, in Chinese jiaotou, few SSR markers have been developed, which has been a major obstacle to genetic and breeding studies of the crop.
In the present study, a total of 1494 EST-derived SSR markers were developed, and 100 SSR primer pairs were used to evaluate the quality of the EST–SSR primers. The result showed that 95% primer pairs were successful in amplifying their target sequences. Because these 100 SSRs were randomly chosen from 1494 markers for quality evaluating, the 95% success observed for the amplification could be extrapolated to mean that 1494 markers can be successfully amplified using PCR. The remaining 5% of the SSR primer pairs probably failed either because the primers were designed across splice sites or because large introns were present in the target sequences (Cloutier et al., Reference Cloutier, Niu, Datla and Duguid2009).
The 31-kb interval of the Chinese jiaotou EST–SSRs suggests that EST–SSRs are less prevalent in Chinese jiaotou than in many other plant species (Liu et al., Reference Liu, Zhu, Fu, Tang, Yu, Chen, Luan, Wang and Tang2013b; Ding et al., Reference Ding, Jia, Luo, Zhang, Cong, Liu and Bai2015; Zhang et al., Reference Zhang, Li, Tao, Fang and Qi2015; Hou et al., Reference Hou, Feng and Wu2017) but more prevalent than in garlic (Liu et al., Reference Liu, Zeng, Zhu, Chen, Tang, Mei and Tang2015a). Meanwhile, the trinucleotide motif was the most abundant, which is similar to that patterns observed in other plant species (Liu et al., Reference Liu, Zhu, Fu, Tang, Yu, Chen, Luan, Wang and Tang2013b; Guo et al., Reference Guo, Mao, Cai, Wang, Wu and Qiu2014; Zhai et al., Reference Zhai, Xu, Wang, Cheng, Chen, Gong and Liu2014; Ding et al., Reference Ding, Jia, Luo, Zhang, Cong, Liu and Bai2015; Zhang et al., Reference Zhang, Li, Tao, Fang and Qi2015). However, the number of EST–SSRs, the average distance between EST–SSRs, and the abundance of motifs are all highly dependent on the use of different SSR search criteria and the size of databases (Aggarwal et al., Reference Aggarwal, Hendre, Varshney, Bhat, Krishnakumar and Singh2007). Taken the abundance of motifs as an example, SSR search required more than seven repeats for dinucleotide, whereas for trinucleotide, at least five repeats were demanded, resulting in more dinucleotide motifs being missed than trinucleotides. Therefore, the feature of SSRs presented in this study, such as the average distance of EST–SSRs and the abundance of motifs, only indicated the characterization of these 1494 markers developed, but not all EST–SSRs existed in the transcriptome of Chinese jiaotou.
Previous studies have found that GC-rich SSR motifs are more prevalent in ESTs from monocots than those from dicots (Liu et al., Reference Liu, Zhu, Fu, Tang, Yu, Chen, Luan, Wang and Tang2013b), and the abundance of CCG/CGG motifs is reportedly a specific feature of monocot genomes (Peng and Lapitan, Reference Peng and Lapitan2005). However, in Chinese jiaotou, GAA/TTC motifs were the most abundant, and few SSRs with GC-rich motifs were identified in the 121,008 assembled ESTs. In addition to Chinese jiaotou, relatively fewer GC-rich motifs are also present in garlic, which is another Allium species in which EST–SSRs have been developed in a large scale (Liu et al., Reference Liu, Zeng, Zhu, Chen, Tang, Mei and Tang2015a). Probably, GC-rich SSR motifs are less prevalent in the ESTs of Allium species than in other monocots.
Potential application in breeding of Chinese jiaotou
SSR variations in the expressed sequences might have an important influence on gene function (Li et al., Reference Li, Korol, Fahima and Nevo2004). For example, SSR expansions or contractions in the CDS region are likely to cause frame shifts or deletion/insertion of protein-encoding sequence, which can disrupt the function of the protein encoded by the SSR-containing gene; changes in the length of SSR in 5′ UTRs could regulate gene expression and SSR expansions in 3′ UTRs may cause transcription slippage by affecting transcription and translation. In this study, 1494 SSRs were developed from the 1459 ESTs of Chinese jiaotou, and 624 fell into the CDS, and 589 into UTR, respectively. In addition, among these 1459 SSR-containing ESTs, most of them were annotated with important function, including at least 93 transcription factor-encoding genes. GO functional classification revealed that several categories involved in transcription regulation showed significant enrichment by these EST-containing sequences. Because transcription factor have central roles in regulating plant growth, development, and stress response (Liu et al., Reference Liu, Zhu, Tang, Yu and Tang2013c; Zhu et al., Reference Zhu, Tang, Tang and Liu2014; Zheng et al., Reference Zheng, Zhu, Tang and Liu2016b), these SSR markers will therefore be useful for selecting and pyramiding agriculturally valuable alleles in molecular marker-assisted selection breeding of Chinese jiaotou.
Cross-species transferability of the developed SSR markers
The interspecies transferability of 100 EST–SSRs from Chinese jiaotou to six other Allium species indicated that 97 SSRs possessed cross-species transferability, and 69 of these SSRs exhibited transferability to all six other Allium species. This high transferability is consistent with previous reports in other plant groups (Zheng et al., Reference Zheng, Pan, Diao, You, Yang and Hu2013; Wang et al., Reference Wang, Jiang, Chen, Qi, Peng, Li, Song, Guan, Fang, Liao and Chen2013; Guo et al., Reference Guo, Mao, Cai, Wang, Wu and Qiu2014). In Allium, the high transferability of garlic SSRs to other Allium species, including to Chinese jiaotou, has also been observed (Liu et al., Reference Liu, Zeng, Zhu, Chen, Tang, Mei and Tang2015a). In fact, ~77.5% garlic SSRs primer sets were able to amplify PCR products in Chinese jiaotou (Liu et al., Reference Liu, Zeng, Zhu, Chen, Tang, Mei and Tang2015a); whereas 88% of the primer sets developed in the present study were able to amplify PCR products in garlic. This high transferability suggests that Allium species are closely related. Interestingly, because the Chinese jiaotou SSRs derived from the ESTs that enriched into the GO categories involved in transcription regulation, oxidation–reduction, transport, etc. the high transferability of these SSRs indicated that these GO function categories likely are conserved in Allium species.
New insight into the origin of Chinese jiaotou
Chinese jiaotou is thought to have originated in China and has been cultivated there for over 3000 years (Xu et al., Reference Xu, Um, Kim, Lu, Guo, Liu, Bah and Mao2008). However, the exact region from which the crop originated has yet to be determined. In the present study, 19 local varieties were collected from eight provinces, which are major production regions for Chinese jiaotou, and four wild genotypes were collected from three provinces. Analysis of the genetic relationships among the 23 accessions indicated that two of the cultivated varieties, which originated from northern Hunan, are closely related to the four wild genotypes. Therefore, our results provide a new clue for understanding the origin of Chinese jiaotou.
In addition, seven of the accessions, including the four wild genotypes, produce round bulbs, whereas the other 16 produce elliptical bulbs. Interestingly, all six accessions in cluster II produce round bulbs, and all 16 that produce elliptical bulbs were included in cluster I and II. Thus, the SSR-based classification of the 23 accessions was similar to that based on bulb shape. Furthermore, the topology of our phylogenetic tree suggests that round bulb shape is ancestral and that elliptical bulbs only arose after domestication. Therefore, it is possible that bulb shape was an important trait during the early domestication of Chinese jiaotou.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1479262117000338
Acknowledgements
This work was supported by grants from The Agricultural Science and Technology Innovation Program of China (CAAS-ASTIP-IBFC).