INTRODUCTION
Pathogenic Leishmania are protozoan parasites responsible for a spectrum of human diseases collectively called leishmaniases (Pearson and Sousa, 1996). The wide range of pathologies depends not only on the infecting species but also on a complex host-parasite interaction. The availability of the Leishmania major genome sequence has provided a unique insight into the content and organization of the parasite's genetic information (Ivens et al. 2005). Such a comprehensive view of its genome and our ability to genetically manipulate this trypanosomatid constitute a fertile ground for the development of functional studies, such as the generation of mutant collections of the parasite.
Leishmania is mostly diploid and is believed to have an asexual life-cycle (Iovannisci et al. 1984). These features pose a major challenge to the generation of homozygous mutants, especially when dealing with phenotypes that cannot be easily rescued. Attempts to inactivate genes that are essential for this parasite invariably induce DNA amplification and/or changes in ploidy, revealing a significant genome plasticity (Cruz et al. 1993; Dumas et al. 1997). Targeted gene replacement is widely used to obtain Leishmania mutants (Cruz and Beverley, 1990) and may involve several cloning steps to produce the adequate reagents. Moreover, the generation of null mutants requires consecutive rounds of targeting with independent selectable markers (Cruz et al. 1993; Dumas et al. 1997; Zhang et al. 2003). Alternatively, loss of heterozygosity (LOH), which was described for a few loci (Gueiros-Filho and Beverley, 1996; Pedrosa and Cruz, 2002), can be achieved after subjecting heterozygous cell lines of the parasite to increased concentrations of their selective antibiotic.
Transposon-based mutagenesis constitutes a useful approach not only to the introduction and/or the inactivation of phenotypes, but also to the study of the relationship between gene structure and function (Hamer et al. 2001b). A productive transposition event can be generated either within the target organism or by shuttle mutagenesis. In the latter case, the transposon is mobilized in vivo or in vitro and then introduced into the target organism aiming at a phenotypic interference (Ross-Macdonald et al. 1999; Hamer et al. 2001a).
The Drosophila mariner Mos1 transposon belongs to the Tc1/mariner superfamily of transposable elements, which are effective instruments for gene-delivery strategies (Plasterk et al. 1999; Coates et al. 2000; Williams et al. 2005). The malleability of the Mos1 element allowed the establishment of an in vitro transposition system (Tosi and Beverley, 2000) and the design of a toolkit for shuttle mutagenesis in this parasite (Goyard et al. 2001; Robinson et al. 2004). Modified Mos1 elements are a reliable tool for gene trapping, primer-island sequencing approaches (Pedrosa et al. 2001; Augusto et al. 2004) and shuttle mutagenesis protocols (Marchini et al. 2003). This transposon can also mobilize in vivo within Leishmania (Gueiros-Filho and Beverley, 1997) and Trypanosoma genomes (Leal et al. 2004).
Herein we report the use of in vitro shuttle mutagenesis as a source of reagents for targeted disruption of the putative second-largest subunit of RNA Polymerase III (RPC2), a telomere-located Leishmania gene. The specific integration of the disruption reagent indicates that this approach is an efficient and rapid means of producing mutant parasites. Moreover, the results presented here reveal new features of this parasite's ability to promote genome rearrangements during the targeting of an essential gene. The functional complementation of the targeted gene allowed the further inactivation of alleles and generated a wide range of reagents that are adequate for the study of the mechanisms leading to the observed genome plasticity. The preservation of RPC2 was not mediated by an increase in number of copies or by ectopic insertion into other chromosomes, which suggests that the genomic plasticity of this parasite is not restricted to the current models of DNA rearrangement and amplification (Beverley, 1991; Genest et al. 2005).
MATERIALS AND METHODS
Leishmania cultures and transfection
Promastigote forms of L. major LT252 (MHOM/IR/1983/IR) were grown at 26 °C in M199 medium supplemented with 10% heat-inactivated fetal calf serum (Kapler et al. 1990). Transfectants from CC1, a LT252 clonal line, were selected on M199-agar plates with 32 μgċml−1 of hygromycin B (Gibco BRL) and/or 40 μgċml−1 of nourseothricin (Werner Bioagents) as required. Individual clones were transferred to liquid M199 medium supplemented with 16 μgċml−1 of hygromycin B, and/or 20 μgċml−1 of nourseothricin. Promastigote cells (8·106·ml−1) were pelleted at 2000 g for 10 min at 4 °C for the preparation of genomic DNA or RNA extraction and transfection assays. The electroporation protocol used has been previously described (Kapler et al. 1990). Mutant parasites were named according to the nomenclature of Clayton et al. (1998).
DNA and RNA manipulation
Large-scale plasmid or cosmid DNA preparation was carried out using alkaline lysis followed by caesium chloride gradient (Sambrook et al. 1989). Total DNA from promastigotes was extracted as previously described (Tosi et al. 1997). Bal31 assays used 2U of the enzyme (Biolabs) for different incubation periods; the reaction was stopped by adding EGTA to a final concentration of 20 μM and samples were digested with the restriction enzyme of choice. Intact chromosomes were prepared in agarose blocks (Cruz and Beverley, 1990) and resolved by Pulse Field Gel Eletrophoresis (PFGE) using a Bio-Rad CHEF Mapper apparatus. The PFGE separation of episomal DNA used 10-sec constant pulses for 18 h. Total RNA from promastigotes was extracted using TRIzol® (Gibco BRL). Aliquots containing 15 μg of total RNA were equilibrated in formamide, formaldehyde and MOPS buffer, heated for 15 min at 65 °C and separated in a 1·5% agarose formaldehyde/MOPS gel (Sambrook et al. 1989). DNA and RNA transfer to nylon membranes, hybridization and washing conditions were conducted as previously described (Tosi et al. 1997). Radio-isotope labelled probes were prepared using the random priming method (Feinberg and Vogelstein, 1983). Membranes were exposed to a Kodak Diagnostic Film or to a Molecular Dynamics Storage Phosphor Screen for densitometric analysis. Polymerase Chain Reaction (PCR) and Reverse transcriptase-PCR (RT-PCR) reactions were performed with Ready-To-Go™ PCR and RT-PCR Beads (Amersham Pharmacia Biotech) according to the manufacturer's specifications using the primers (a) 5′-TAGAAGTCGCGCTCGTCAA-3′; (b) 5′-TTCCTCGCCAACGTGCTACT-3′; (c) 5′-GAACGCGTGGATCCTCTAG-3′; (d) 5′-TAGTGCCCACTTTTTCGCAA-3′; (e) 5′-TTGCTATGGCGCATTCATC-3′; (f) 5′-TGCCCTGTAGCTCTTGGATAA-3′ and (g) 5′-TCTCATCAGCTCCGACATGTT-3′.
Molecular constructs
Escherichia coli strains DH10B (Gibco BRL) and DH5α-λpir (Garraway et al. 1997) were used in this study. All strains were grown in LB medium supplemented with the appropriate drug (100 μgċml−1 ampicillin (Gibco BRL); 50 μgċml−1 hygromycin B; or 50 μgċml−1 nourseothricin). Bacteria were transformed by electroporation or heat-shock of Ca2+-prepared cells.
The target for transposition was a 4·3 kb HindIII fragment (HH4·3) subcloned from cosmid E08 (GeneBank Accession number AH010590) (Pedrosa et al. 2001), which bears 36 kb of the chromosome 20 subtelomeric end. The construction of transposon ELSAT (1020 bp) included the PCR amplification of the SAT cassette (the AG sequence, the EM7 promoter and the streptotrycin acetyl transferase gene) using the primers LT22 (5′-CGCTCTAGAGGATCCAGGCGTTCGAA-3′) and LT23 (5′-CGCAGATCTAGATCTCCGAGGCCTG-3′) and pELSAT as template (Garraway et al. 1997). The PCR product was digested with XbaI and inserted into the XbaI site of pELHY6Δ-0 (Garraway et al. 1997). The plasmid or cosmid DNA templates bearing the transposon insertion were prepared using a modified alkaline lysis method (Sambrook et al. 1989) in a 96-well format, followed by a purification step with 96-well MultiScreen filter plates (Millipore). Sequencing reactions used the Big Dye terminator chemistry with a specific primer for each transposon. The primers for ELSAT primer island sequencing were 5′-GAACGCGTGGATCCTCTAG-3′ (LT25). Single-pass sequencing was carried out on ABI3100 sequencing machine (Applied Biosystems).
Transposase purification and transposition assay
The recombinant Mos1 transposase preparation and the transposition reactions were performed as previously described (Tosi and Beverley, 2000). The reaction products were recovered following transformation into E. coli which lacked the pir gene product.
RESULTS
Shuttle mutagenesis
The target used for disruption by shuttle mutagenesis is presented in Fig. 1A. The RPC2 gene (LmjF20.0010) is the nearest to one of the telomeric ends of L. major (LT252) chromosome 20 (Pedrosa et al. 2001). As shown in the figure, the fragment HH4·3 used as target for transposition contains 92% of the predicted RPC2. Sequencing of cosmid E08 confirmed that the RPC2 gene did not present any noticeable alteration, as revealed by sequence alignment of the episomal and genomic copies of the gene (data not shown).

Fig. 1. Shuttle mutagenesis of the telomere-located RPC2 gene. (A) Schematic representation of Leishmania major chromosome 20 extremity contained in cosmid E08. As indicated, the 4·3 kb HindIII fragment was used as target for in vitro transposition. Vertical lines across RPC2 gene represent mapped insertions irrespective of the direction of the transposon. The event of interest H01 is marked with a closed triangle. RP1 and RP2 represent the probes used in Southern and Northern experiments; arrows a, b, f and g (out of scale) indicate location and direction of primers used in PCR or RT-PCR protocols; H, HindIII; Sp, SphI; Sc, SacI; B, BstEII; N, NarI; E, EcoRV; P, PvuII; S, SalI; K, KpnI. (B) Schematic representation of the disruption fragment H01∧rpc2::SAT containing the modified Mos1 transposon ELSAT. The grey arrows represent the selectable resistance marker streptothricin acetyl transferase gene (SATR); 5′IR, mariner 5′ inverted repeat; 3′IR, mariner 3′ inverted repeat; AG, Leishmania trans-splicing acceptor site; arrowheads upstream of resistance genes represent Escherichia coli promoter sequence; SAT represents the probe used in Southern and Northern experiments; arrows c, d and e (out of scale) indicate location and direction of primers used in PCR or RT-PCR protocols.
The transposon used in the disruption protocol is shown in Fig. 1B. The construction of transposon ELSAT and its potential application in disruption strategies have been described elsewhere (Goyard et al. 2001). The mobile element ELSAT contains a shuttle cassette encoding the streptotrycin acetyl transferase gene (SAT). The efficiency of in vitro transposition of ELSAT into different targets is comparable to that of modified Mos1 transposons (Tosi and Beverley, 2000). The insertion events across fragment HH4·3 are indicated in Fig. 1A and were identified by restriction mapping and/or primer island sequencing. As previously reported for other Mos1 transposons (Augusto et al. 2004), the insertion of these elements showed little regional specificity or orientation bias (P>0·05; χ2 test). The selected insertion event used to generate the linear fragment for target disruption of RPC2 is indicated in Fig. 1A as H01. The major features of the disruption fragment H01∧rpc2::SAT are shown in Fig. 1B. Varying amounts of the fragment, which contains the selectable marker flanked by 0·8 and 1·2 kb of the target sequence, were transfected into the parasite.
Targeted disruption of RPC2 gene
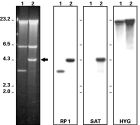
A wild-type cell line (L. major LT252) was transfected with the targeting fragment H01∧rpc2: SAT to generate the disruption of a single RPC2 allele. Clones were selected in medium containing nourseothricin. Two transfection experiments using 2 and 5 μg of the gel-purified targeting fragment resulted in the selection of 2 and 4 clones, respectively. The targeted disruption was confirmed by Southern analysis of EcoRV-digested genomic DNA from these 6 clones using fragment RP1, RP2 and SAT probes (probe location is depicted in Fig. 1A and B). Fig. 2A shows that probe RP1 revealed the expected 4·3 kb fragment from the disrupted gene and a wild-type 3·3 kb fragment from the intact loci for clones 1 and 2. The SAT probe detected the 4·3 kb in both clones. The use of probe RP2, which detected not only the EcoRV fragment bearing the integration but also the telomere-proximal EcoRV fragment, suggested the overall integrity of the locus after targeted disruption (Fig. 2A). Hybridization of PFGE-separated chromosomal DNA revealed the presence of the SAT marker on a ~760 kb chromosome in the selected heterozygous clones (Fig. 2B). In addition, the RP1 probe detected the equivalent chromosomal band in the wild-type cell line and in clones HZ.1 and HZ.2 (RPC2/∧rpc2:: SAT.1 and .2). The specific integration of the disrupted locus was further confirmed in Bal31 nuclease assays. As shown in Fig. 2C, the exonuclease activity on genomic DNA from mutant HZ.2 caused a progressive shortening of the SalI fragment, which bears the RPC2 gene and the SAT marker at one of the chromosomal ends. The use of an internal probe, which detects a 260-nt stretch downstream of the DHFR-TS gene in chromosome 6, confirmed the specificity of the Bal31 nuclease activity and is indicated with an asterisk in Fig. 2C. The targeted disruption of RPC2 was also demonstrated using PCR with different primer sets (presented in Fig. 1A) and clone HZ.2 genomic DNA as a template. Amplification of truncated RPC2 (primer sets a/b and a/c) and SAT (primer set d/e) was only detected in the disrupted cell line (Fig. 2D). The integration of the SAT marker in the genome of selected mutants was maintained even after removal of nourseothricin selective pressure for more than 300 generations (data not shown). Table 1 summarizes the characteristics of clone HZ and specific mutants used in this work.

Fig. 2. Specific integration of the fragment H01∧RPC2: SAT into the genome. (A) Southern experiments from the wild-type (WT) and mutant clones HZ.1 (1) and HZ.2 (2), derived from 2 distinct transfection experiments of the targeting fragment; Southern blots of EcoRV-digested genomic DNA were probed with fragments RP1, RP2 and SAT (refer to Fig. 1). (B) The same probes were used in blots of PFG-separated chromosomes from parental and mutant cell lines. (C) Time-course digestion of clone HZ.2 genomic DNA with exonuclease Bal31; samples were further digested with SalI and analysed in Southern blots; signal of a 260-nt internal probe, located downstream of the DHFR-TS gene in chromosome 6, is marked with an asterisk and confirmed the specificity Bal31 activity. (D) The targeted disruption of the RPC2 was further confirmed by PCR of WT and HZ.2 genomic DNA using different sets of primers (refer to Fig. 1 for location of primers a, b, c, d, e, f and g); the arrowhead and the arrow indicate the amplification of 1160 and 160 bp-sized fragments, respectively.


Fig. 3. Attempt to generate a RPC2 null mutant. Southern analysis of EcoRV-digested genomic DNA (A) and PFG-separated chromosomes (B) of parental cell line (WT) and mutants HZ.1400 and HZ.2400 (1 and 2, respectively), which were selected after culturing the heterozygous cell line in 400 μgċml−1 nourseothricin. Southern blots were probed with fragment RP1.

Fig. 4. Genomic variability at the RPC2 locus after selective pressure. Southern analysis of KpnI-digested genomic DNA (A) and (B) and PFG-separated chromosomes (C) from cell lines HZ400[E0816] and HZ0. The HZ400[E0816] mutants were obtained after transfection of cosmid E08 into the heterozygous cell line HZ followed by culturing in nourseothricin 400 μgċml−1. HZ0 clones were isolated after culturing the mutants HZ400[E0816] without drug pressure for 200 generations to eliminate the circular episome. The HZ400[E0816] mutants shown in lanes 4, 5 and 6 were cloned from the cell line shown in lane 2. Likewise, HZ0 clones in lanes 1, 2, 3, 4, 6 and 7 originated from clone in lane 5. The open circles indicate fragments bearing undisrupted copies of RPC2; the asterisks indicate the episomal elements. The electrophoresis protocol that allowed the separation seen in A and B used a 0·5% agarose gel; 45 Vċcm−1 for 40 h. Mutant nomenclature is: HZ400[E0816] (RPC2/ ∧rpc2::SAT400 [cLHYG16 E08∧rpc2::SAT]); HZ0 (∧rpc2::SAT0/ RPC2).
Further inactivation of RPC2 copies
The generation of a RPC2 null mutant was attempted by inducing the LOH, an approach previously used in Leishmania (Gueiros-Filho and Beverley, 1996; Pedrosa and Cruz, 2002). The heterozygous cell line was cultured in 400 μgċml−1 of nourseothricin in order to disrupt other copies of RPC2. This strategy did not lead to the generation of a RPC2 null mutant, which indicates that RPC2 is an essential gene. Southern analysis of EcoRV-digested genomic DNA from two selected clones revealed the preservation of an intact RPC2 locus (Fig. 3A). Hybridization of PFGE-separated chromosomal DNA from these clones also showed an amplification of the SAT marker. As seen in Fig. 3B, one of the selected clones (HZ.1400) carried an amplified SAT-containing episome, while the other (HZ.2400) seemed to present an altered ploidy.
As described for another Leishmania gene (Tovar et al. 1998), we expected chromosomal null mutants of the RPC2 gene to be generated after functional complementation. Therefore, the cosmid bearing the intact RPC2 gene was transfected into the heterozygous cell line (HZ.1 clone shown in Fig. 2). Following transfection of cosmid E08, selected clones were cultured in 400 μgċml−1 of nourseothricin for 100 generations. Cell lines selected under this condition were named HZ400[E0816]. The resulting disruption events were tested using Southern analysis of KpnI-digested genomic DNA. The endonuclease KpnI cleaves upstream of RPC2 and releases the end of chromosome 20 (Fig. 1A). This restriction pattern allows the discrimination between the episomal and the wild-type chromosomal RPC2 loci (~20 kb and ~9·7 kb fragments, respectively). As seen in Fig. 4A, Southern analysis using RP1 and SAT probes revealed that the heterozygous mutant HZ bears at least 2 intact copies of RPC2, in addition to the targeted copy. These results suggest the existence of at least 3 copies of this gene in the parental cell line LT252. The distinction among these copies was not possible in regular electrophoresis conditions, as seen in Fig. 2C, and required a different electrophoresis protocol, as indicated in Fig. 4 legend.
Southern analysis of HZ400[E0816] clones using the RP1 probe detected KpnI fragments that varied in size from ~7 to more than 20 kb, in contrast to the wild-type cell line. Such fragments, indicated by open circles in Fig. 4A and B, were not detected by the SAT probe, which suggests they were undisrupted copies of RPC2. Moreover, the intensity of the hybridization signals of these new KpnI fragments does not indicate DNA amplification of the locus. The preservation of undisrupted chromosomal copies of RPC2 in HZ400 [E0816] clones was also confirmed by the loss of cosmid E08 in the absence of hygromycin B selective pressure. As seen in Fig. 4, Southern analysis of PFG-separated chromosomes and KpnI-digested genomic DNA from hygromycin B-sensitive (HZ0 clones) with RP1 and SAT probes revealed the loss of the episomal molecule, which is indicated by an asterisk in Fig. 4A, B and C. It is noteworthy that the fragments of different sizes bearing intact copies of RPC2 were maintained in HZ0 clones. In spite of the size variation of KpnI fragments, hybridization of the RP1 probe to EcoRV-digested DNA from these heterozygous clones revealed a constant pattern for both wild-type and disrupted loci (data not shown). Hybridization signals of RP1 and SAT probes on chromosomal blots of HZ0 clones were detected exclusively in chromosome 20 (Fig. 4C), indicating the absence of ectopic rearrangements of the targeted locus. These results suggest that the appearance of new KpnI fragments containing the undisrupted RPC2 gene was not mediated by chromosomal translocation or DNA amplification as previously described for other loci (Genest et al. 2005).
Another striking feature revealed during the attempts to generate a RPC2 chromosomal null mutant was the rearrangement of the SAT gene into the episomal molecule E08. As seen in Fig. 4A and B, the clones HZ400 [E0816], which were selected in high doses of nourseothricin, bear an episome (indicated as an asterisk) that is detected by both RP1 and SAT probes. The integration of a marker originally present in chromosome 20 of the heterozygous mutant HZ into cosmid E08 was confirmed in episomal molecules recovered from HZ400 [E0816] clones. Fig. 5 shows the comparison between the cosmid E08 before transfection and after rescuing from HZ400 [E0816] clones. The only noticeable alteration in their EcoRV restriction pattern is the increase in size of the 3·3 kb fragment containing the wild-type RPC2 locus. Southern blots hybridized with the RP1 and the SAT probes confirmed that the rescued molecule carried the disrupted locus (Fig. 5). In order to confirm that the rescued episome was not an amplicon generated in HZ400 [E0816] clones, the same blot was hybridized to a HYG probe, which detected the cosmid backbone, the cLHYG vector (Fig. 5).

Fig. 5. Targeting of the RPC2 locus within cosmid E08. EcoRV restriction pattern and Southern analysis of cosmid E08 before transfection into the parasite (lane 1) and after rescuing from clone HZ400[E0816] (lane 2). The probes used in Southern blot hybridization were RP1, SAT and HYG, which detected the cosmid backbone.
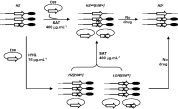
As shown here and reported elsewhere for other loci (Dumas et al. 1997), transfection of the intact gene into a heterozygous clone, followed by the increase in drug concentration, did not rescue null mutants. Surprisingly, the disruption of chromosomal copies of the RPC2 locus was selected in the absence of high nourseothricin selective pressure after transfection of cosmid E08 into heterozygous clones. Southern analysis of KpnI-digested DNA from representative clones suggested the disruption of the RPC2 alleles (Fig. 6). In half of the selected clones the RP1-positive KpnI fragments of chromosomal origin also hybridized with the SAT probe (clone LOH [E0816]; Fig. 6). The other half of the mutants (clone HZ [E0816]; Fig. 6) carried at least 1 intact RPC2 locus and an untargeted cosmid E08. The integration of the SAT marker into the episomal molecule was also observed in mutant LOH [E0816]. As seen in Fig. 6, this clone bears an additional KpnI fragment of ~21 kb that is detected not only by the RP1, but also by the SAT probe, which indicates that some of the episomal molecules carry a disrupted version of RPC2. Based on these results and on our ability to dilute cosmid E08, Fig. 7 shows a schematic representation of the possible configuration of RPC2 alleles and episomal molecules in clones selected during the attempts to generate a RPC2 homozygous mutant.

Fig. 6. Further disruption of RPC2 copies. Southern analysis of KpnI-digested genomic DNA from the heterozygous mutant HZ and cell lines HZ [E0816] (lane 1) and LOH [E0816] (lane 2) selected exclusively in hygromycin B after the transfection of cosmid E08 into the HZ.2 mutant. Southern blots were probed with fragments RP1 and SAT.

Fig. 7. Schematic representation of the possible configuration of RPC2 mutants. Configuration of alleles and episomal molecules in the heterozygous mutant HZ and mutant lines selected during the attempts to generate a RPC2 knockout. The complete mutant nomenclature is: HZ (RPC2/RPC2/∧rpc2::SAT20); HZ [E0816] (RPC2/∧rpc2::SAT/ ∧rpc2::SAT [cLHYG16 E08 RPC2].1); LOH [E0816] (∧rpc2::SATLOH [cLHYG16 E08 RPC2/ ∧rpc2::SAT]) and HZ400[E0816] (RPC2/∧rpc2::SAT400 [cLHYG16 E08 rpc2::SAT]).
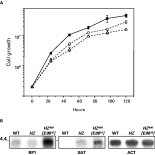
The heterozygous cell line and the mutants presented in Fig. 4A and Fig. 6 had a compromised growth when compared to wild-type cells and entered the stationary phase at a significantly (P<0·05) lower density (Fig. 8A). This effect was not observed in wild-type cells transfected with the cosmid E08, which indicates that the phenotype was not caused by the over-expression of the circular intact loci (data not shown). Northern blots using total RNA isolated from wild-type and mutant cell lines were probed with RP1, SAT and actin gene (Fig. 8B). Densitometry analysis of the data showed no significant difference in the level of RPC2 transcript between the heterozygous cell line and the wild-type strain. However, mutant HZ400[E0816] had significantly higher levels of both RPC2 and SAT transcripts.

Fig. 8. Phenotypic analyses of RPC2 mutants. (A) Cell growth pattern of wild-type (closed square), the heterozygous mutant HZ (open circle) and cell line LOH [E0816] (open triangle) cultivated in drug-free medium. (B) Northern blot analysis of total RNA isolated from wild-type (WT), mutant HZ and cell line HZ400 [E0816]; fragments RP1, SAT and the actin gene were used as probes.
DISCUSSION
The availability of Leishmania mutants has greatly contributed to extend our knowledge of this parasite's biology (Turco et al. 2001). In this work we have used a shuttle mutagenesis approach to target a telomere-located essential gene of L. major. The use of the mariner in vitro transposition system led to the generation of collections of reagents adequate for targeted integration into the parasite's genome. The availability of a straightforward strategy for mutant generation is particularly relevant if we take into account that more than half of the predicted Leishmania genes code for hypothetical proteins (Ivens et al. 2005).
The integration efficiency into the L. major chromosome 20 subtelomeric region was comparable to that previously reported for L. major CC1 strain (Cruz and Beverley, 1990; Cruz et al. 1991). Although transcript levels of the targeted RPC2 gene were not significantly reduced in the heterozygous mutant, a low growth was observed, confirming that first-round disruption mutants may be important tools for gene dosage studies, as already demonstrated for L. major and L. donovani Trypanothione reductase (Dumas et al. 1997). Moreover, a cell line bearing a selectable marker at the telomere can be used in the study of the expression of telomere-located Leishmania genes.
The generation of null mutants of Leishmania requires successive and independent events of gene inactivation. This can be achieved by rounds of targeting or by one targeted disruption combined with LOH, which uses high doses of antibiotics to select a homozygous cell line and has the advantage of saving selectable markers for targeted disruption of other loci or for functional complementation protocols (Gueiros-Filho and Beverley, 1996). Instead of leading to a RPC2 null mutant, the use of high doses of nourseothricin selected parasites with multiple copies of the targeted locus, providing evidence that this subunit of the Leishmania RNA Polymerase III complex is essential for the parasite. Although null mutants are readily generated for non-essential genes in Leishmania, attempts to inactivate vital genes may lead to extensive karyotype alterations, which ensure the preservation of the targeted gene (Cruz et al. 1993; Tovar et al. 1998; Genest et al. 2005).
The supplementation with either an episome carrying the intact locus or the implicated metabolite can be enough to guarantee the replacement of chromosomal copies of an essential gene (Gueiros-Filho and Beverley, 1996; Tovar et al. 1998). Although RPC2 transcript levels were elevated in cell lines complemented with the intact gene, the generation of chromosomal null mutants of RPC2 was not possible even in the presence of high doses of the selective drug. The genetic variability, which granted the preservation of intact copies of RPC2 in the selected mutants, did not involve chromosome translocations or the generation of amplicons as widely documented in Leishmania (Beverley, 1991; Genest et al. 2005). The different KpnI fragments containing the intact RPC2 locus may represent an unexpected variation in the size of chromosome 20 subtelomeric region in these mutants. Size variation between chromosome homologues has already been described in Leishmania and may involve gene amplification and/or rearrangement of subtelomeric repetitive sequences (Sunkin et al. 2000). Leishmania telomeres contain varying amounts of typical subtelomeric repeats, such as LST-R (Myler et al. 1999; El-Sayed et al. 2005); LCTAS (Fu and Barker, 1998) and LST-R533 (Pedrosa et al. 2006). Therefore, the variability observed in the mutant parasites described in this work may have been mediated by repetitive elements LCTAS and LST-R533, which are present at the subtelomeric region of L. major LT252 chromosome 20. Chromosomal ends of different protozoan parasites play relevant roles in virulence and survival within their hosts (Borst and Rudenko, 1994; Freitas-Junior et al. 2000; del Portillo et al. 2001; Horn and Barry, 2005). Subtelomeres of Trypanosoma brucei may contain long tandem arrays of variant surface glycoprotein (VSG) gene sequences and a single VSG expression site, whose control is a key feature of antigenic variation in this parasite (Rudenko et al. 1998). Chromosome size polymorphism in this trypanosomatid is mainly caused by a high degree of variability in subtelomeres, which suggests that it can be partially credited to the VSG system (Melville et al. 1999; Rudenko, 2000; Horn and Barry, 2005). However, the subtelomeric regions of Leishmania do not seem to be involved in such antigenic variation mechanisms and are relatively shorter and poorer in repetitive elements when compared to other pathogenic trypanosomatids (El-Sayed et al. 2005).
The variability described here may provide new information about the genome structure and control of gene expression in Leishmania. The variation forcing the maintenance of the loci in its original chromosomal context suggests that tightly regulated RPC2 expression is crucial for cell homeostasis in L. major. In contrast to the endogenous locus, the selected mutants indicated a marked preference for the preservation of the SAT marker on a supernumerary episome, as revealed by the unexpected targeting of the supplementing circular molecule. The SAT marker, originally present in the chromosome of the heterozygous mutant, was found in the cosmid E08 recovered from different transfectants. DNA recombination between chromosomes and circular episomes is commonly observed in T. brucei (ten Asbroek et al. 1990) and is, in fact, a distinguishing feature of the genetic tools available for this trypanosomatid. On the other hand, Leishmania episomal DNA is fairly stable and rearrangements between episomal elements are seldom reported in this organism (Tobin et al. 1991). Altogether, the mutants and events described in this work may contribute to a better understanding not only of the structure and function of the parasite's telomeres, but also of how their DNA recombination machinery can be used in the control of gene expression.
As discussed above, the mutants generated in this work confirm the genome plasticity of Leishmania and provide new information about the parasite's genome maintenance. In addition, our data suggest that the in vitro shuttle mutagenesis is a reliable source of reagents for targeted disruption protocols in this organism. The diversity of insertion events that can be generated in a single reaction makes this technology more efficient than classical DNA cloning methods (Beverley, 2003). Disruption fragments for several genes can be easily produced, identified and annotated into the genome. A pool of targeting fragments can be introduced into the parasite by a single mass-transfection experiment, allowing the generation of several independent targeted mutants. This is a systematic approach with inherent characteristics of a large-scale post-genomic protocol, as pointed out by Sakamoto et al. (2005). Therefore, replacement events and rescued phenotypes require further and more detailed studies. The mariner toolkit developed for use in this parasite may generate an extensive collection of reagents, which include not only functional knockout fragments but also vehicles for targeted integration of fusion proteins into the genome (Goyard et al. 2001; Robinson et al. 2004; Augusto et al. 2004). Herein we have shown that a simple method for the production of reagents combined with the vast amount of sequence information can greatly facilitate the generation of engineered mutants. A comprehensive collection of mutants will certainly help in the characterization of the parasite's virulence and in the identification of potential targets for the control of leishmaniases.
This work was supported by Fundação de Amparo a Pesquisa do Estado de São Paulo, FAPESP, 98/09805-0, 99/12403-3; and UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases; FMS was sponsored by FAPESP; 01/02527-9. We thank Marlei Josieli Augusto for technical assistance.