


Introduction
Cryopreservation of oocytes is one of the most actively studied topics in the field of assisted reproductive technology. The technology of oocyte cryopreservation could be used to preserve surplus mature oocytes from clinical in vitro fertilization (IVF) cycles. Stored in the oocyte banks, cryopreserved oocytes can be provided to patients with premature ovarian failure (POF), and patients who are undergoing chemotherapy and radiation treatment for malignant diseases (Porcu et al., Reference Porcu, Fabbri, Damiano, Giunchi, Fratto, Ciotti, Venturoli and Flamigni2000). The cryopreservation methods include slow freezing and vitrification. Although the slow-freezing method has been significantly improved in the past decade (Stachecki et al., Reference Stachecki, Cohen and Willadsen1998; Fabbri et al., Reference Fabbri, Porcu, Marsella, Rocchetta, Venturoli and Flamigni2001; Quintans et al., Reference Quintans, Donaldson, Bertolino and Pasqualini2002), this process is generally time consuming and requires expensive equipments (Jain et al., Reference Jain and Paulson2006). Compared with the slow-freezing method, the vitrification method takes less time, is less labor intensive and needs less complex equipments. More importantly, the vitrification method has been proved to be effective in animals and some clinical applications. Therefore, the study of vitrification has attracted much attention recently. Research groups around the world have carried out intensive studies and got some promising results.
Despite the benefits mentioned above, there is not one generally accepted vitrification protocol in clinical practice. The existing vitrification protocols and their outcomes vary a lot (Gook et al., Reference Gook and Edgar2007). The purpose of this study is to assess a vitrification protocol modified from the one used for human embryos in our clinic IVF center (Sun et al., Reference Sun, He, Yu, Deng and Liu2005). The new protocol increased the time of the oocytes in vitrification solution as well as the time for the oocytes to reach the sucrose free stage in the warm up step. We have tested the effectiveness of this protocol by examining the following characteristics of oocytes: the configurations of the oocytes' spindles and chromosomes, the oocytes' DNA fragmentation, and the oocytes' ability to be fertilized and its ability to develop into blastocysts. This study provided some useful ideas and data set in the development of a future standard vitrification protocol in clinical practice.
Materials and Methods
Media
All media used in this research were the sequential culture media (Quinn's advantage protein plus media, Sage Pharmaceuticals, USA) unless stated otherwise.
For in vitro experiments, such as transferring ovaries and tubes, preparing the vitrification solution, we used the HTF–HEPES medium [human tubal fluid(HTF)with 5.0 mg/ml human albumin and HEPES as buffer). The fertilization–HTF medium was used for sperm capacitation and making insemination drops. The cleavage medium and the blastocyst medium were used for embryo culture. Except for HTF–HEPES medium, all other media used bicarbonates as buffer. All meida were prepared in sterile tissue culture dishes one day before use. The culture dishes were then placed in a humidified 37 °C incubator with 5% CO2 to equilibrate overnight. In order to allow adequate gas exchange, all dishes were put in the incubator with their lids taken off.
Oocyte collection
The use of animals in this study was approved by the ethics review board of the hospital.
Oocytes were collected from 4- to 5-week old ICR female mice. They were injected intraperitoneally with 10 IU pregnant mare's gonadotrophin (PMSG, Ningbo City Hormone Co. Ltd) first, and then with 10 IU of human chorionic gonadotrophin (hCG, Ningbo City Hormone Co. Ltd) 48 h later. The mice were killed by cervical vertebra dislocation 13–16 h after hCG, treatment. The oviducts were excised and put in HTF–HEPES medium. In order to collect cumulus–oocyte complexes (COCs), the oviducts were incubated in HTF–HEPES with 80 IU/ml hyaluronidase (SAGE IVF, USA) for about 1 min, the COCs were then denuded from the surrounding granulosa cells. Oocytes were washed twice with fertilization–HTF medium. Mature oocytes with the first polar body were collected and cultured in fertilization–HTF medium in the 5% CO2 incubator at 37 °C for future study.
The oocytes were randomly assigned to the vitrification group and the control group.
Oocyte cryopreservation and warm-up
The cryoloop used for vitrification was a nylon loop mounted on a stainless steel pipe, which was inserted in the lid of a 2 ml cryogenic vial (Hampton Research). A small magnet was mounted on the top surface of the lid (the opposite side of the loop). To move the loop, we used a handle with another small magnet on the tip. The magnetic force between the two pieces of magnets allowed us to make different moves easily and precisely when cryopreserving the oocytes with liquid nitrogen.
The pretreatment solution, vitrification solution, and dilution solutions were all made with HTF–HEPES medium by adding different chemicals. The pretreatment solution contained 7.5% (v/v, 1.06 M) dimethyl sulphoxide (ME2SO, Sigma, USA) and 7.5% (v/v, 1.3 M) ethylene glycol (EG, Sigma, USA). The vitrification solution contained 15% (v/v, 2.1 M) ME2SO, 15% (v/v, 2.6 M) EG, 5.8mg/ml Ficoll 400 (Sigma, USA) and 0.58 M sucrose (Sigma, USA). The five-step dilution solutions contained 1 M, 0.5 M, 0.28 M, 0.17 M and 0 M sucrose, respectively.
All vitrification and dilution process were carried out at room temperature. The pretreatment and vitrification solutions were placed in 4-well culture plates (Nunc, Roskilde, Denmark). Ten oocytes were washed and equilibrated in pretreatment solution for 2 min, and then transferred to vitrification solution and equilibrated. Finally, the oocytes were loaded onto the nylon loop, which had previously been dipped into vitrification solution to create a thin film. This step is very critical: the medium in the film should be as little as possible to allow quick cooling in the next step. For the same reason, the loop containing oocytes should be plunged into liquid nitrogen as quickly as possible. Once the oocytes were transferred into the vitrification solution, they should be plunged into liquid nitrogen within 35–50 s. This small time window would permit complete permeation of the cryoprotectants through the oocytes, yet cause least possible damage to the oocytes because of chemical toxicity and osmotic shock. The loop containing the oocytes was then screwed onto a cryovial that was predipped in liquid nitrogen. The whole vial was quickly put back in a liquid nitrogen tank for storage.
After stored in liquid nitrogen for several days, the oocytes were warmed up and eventually transferred to a sucrose free medium following a five-step dilution procedure. First, the cryogenic vial with oocytes was opened while still in liquid nitrogen. The loop containing oocytes was removed from liquid nitrogen and left in air for about 10 s, and then directly placed into a well filled with HTF–HEPES medium containing 1 M sucrose. The oocytes were immediately expelled into this media from the loop. They were rinsed 4–5 times in the media, removed after 1 min in total, and then transferred to HTF–HEPES containing 0.5 M sucrose, rinsed and equilibrated for 1 min. The sucrose concentration was further decreased by rinsing and equilibrating the oocytes in the following media sequentially: HTF–HEPES/0.28 M sucrose for 2 min, HTF–HEPES/0.17 M sucrose for 3 min, and plain HTF–HEPES for 5 min. After these steps, the oocytes were put back into fertilization–HTF medium in the 37 °C incubator.
Evaluating the vitrified oocytes' survival
The morphological survival rates of thawed oocytes were determined using the following criteria: survived oocytes – the zona pellucida and the membrane of the oocytes were intact with a normal perivitelline space and normal cytoplasm refraction; and dead oocytes were those with membrane rupture, missing perivitelline, missing cytoplasm refraction and/or being granular degenerated.
Immunostaining of the spindles and chromosomes
Only the morphologically survived oocytes in the study group were treated in this step. They were divided into three subgroups based on the incubation time in fertilization–HTF after warmed – 0 h, 1 h and 2 h. There were 20–30 oocytes in each subgroup. The oocytes were fixed and stained using the immunofluorescence method and the procedure was as followed: The oocytes were left in 3.6% paraformaldehyde for 30 min at room temperature, 0.5% Triton X-100–PBS 37 °C for 20 h, 115 M glycine + 1% Triton X-100–PBS for 30 min at room temperature, washed with PBS for 15 min, stained with fluorescein isothiocyanate (FITC, Sigma)-conjugated mouse monoclonal anti-β-tubulin antibodies at room temperature for 90 min, washed with PBS twice, stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma) at room temperature for 10 min, washed with PBS twice. Then the oocytes were mounted on slides using ProLong antifade kit (Eugene, OR) stored in the dark at 4 °C before being checked under the fluorescence confocal scanning laser microscope. The microtubulin of the spindles was stained with FITC, which emitted green fluorescence. The chromosomes were stained with DAPI, which emitted blue fluorescence. When collecting FITC fluorescence, the excitation and emission wave lengths were set at 488 nm and 515–535 nm, respectively. For DAPI, the wave lengths were set at 358 nm and 461 nm, respectively.
Under the microscope, normal spindle should be slightly narrow at both ends while the middle part humped up like a spindle. Injured spindles should show fractured or distorted spindle fibres. The chromosomes of normal oocytes should align on the middle of the spindles (plates), while abnormal oocytes should have some scattered chromosomes outside the plates.
Immunostaining of the fragmented DNA: TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling)
TUNEL is an in situ method for detecting the 3′-OH ends of DNA exposed during the internucleosomal cleavage that occurs during apoptosis. Incorporation of biotinylated dUTP allows detection by immunohistochemical procedures. The labelled apoptotic cells may be visualized by light microscopy.
To test the fragmented DNA, the oocytes were stained with DeadEnd Fluorometric Tunel System (Promega, USA) kit following the manufacture's instruction with some minor modifications. The oocytes were treated as followed: (1) the whole cells were fixed in 4% (v/v) paraformaldehyde solution for 1.5 h at room temperature; (2) the oocytes' membrane was permeabilized when they were treated with 0.2% TritonX-100 for 1 h at room temperature; (3) the fragmented DNA were assessed using the DeadEnd Fluorometric Tunel System (Promega, USA). Fixed oocytes were incubated in TUNEL reaction medium for 1 h at 38.5 °C in the dark. During this step, both ends of the fragmented DNA were labelled with rTdT and fluorescein–dUTP; (4) after the reaction stopped, the oocytes were washed and transferred into 2 mg/ml DAPI and incubated for 10 min at room temperature; and (5) the oocytes were then mounted on slides using ProLong antifade kit. The finished slides were stored in −20 °C.
To obtain a positive control, fixed oocytes were treated with 6 U/ml RNase-free DNase I (Promega, USA) for 30 min at room temperature in the dark before the TUNEL reaction, i.e. step 3. The negative control was oocytes treated with solutions that had no rTdT enzymes at step 3.
In vitro fertilization and embryo culture
Spermatozoa were aspirated from the epididymis of 10− to 12−week-old ICR male mice into the fertilization medium. The spermatozoa were capacitated for 1–2 h before insemination. The oocytes were placed in drops of 100 μl fertilization medium and incubated in the 37 °C 5% CO2 incubator. They were then inseminated with approximately 1–2 × 106/ml spermatozoa in each drop of fertilization medium. The fresh oocytes in the control group were immediately fertilized when the spermatozoa got capacitated. After 4–6 h incubation with the spermatozoa, the oocytes were washed several times in the cleavage medium and incubated in 50 μl drops of this kind medium at 37 °C in the 5% CO2 incubator. At this time, the fertilized oocytes had two pronuclei. Successful fertilization was confirmed the next morning if 2-cell embryos were observed. After another 48 h culture, the embryos were washed and then cultured in blastocyst medium to let the embryos grow into blastocyst stage.
Statistical analysis
We performed the chi-squared test and Fisher's Exact tests on the whole dataset with SPSS12.0. p-values <0.05 were considered significantly different.
Results
Spindle and chromosome configurations of the oocytes
Our study included 612 cryopreserved mature oocytes in total. Among them, 601 survived and the morphological survival rate was 98.2%.
The observed abnormal spindles or scattered chromosomes were all minor injuries. No major defects were found in either the study group or the control group (Fig. 1a, b). The numbers of oocytes with normal spindle and chromosome configurations between these two groups were not statistically different (Table 1).
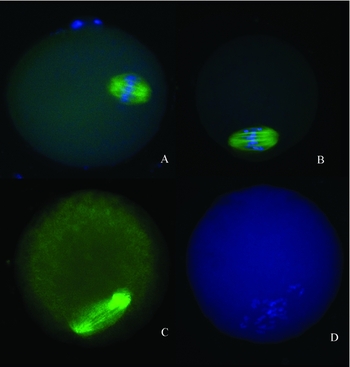
Figure 1 The spindle and chromosome configuration of the thawed oocytes. (a) An oocyte with a normal spindle and chromosome configuration; (b) an oocyte with slightly abnormal spindles, some fractured fibres, and slightly scattered chromosomes; (c) an oocyte with severely abnormal spindles; and (d) the same oocyte as in Fig. 1c., It has severely scattered chromosomes.
Table 1 The spindle and chromosome configurations of oocytes in different groups.

a The cryoloop had too much medium when it was dipped into nitrogen.
b The oocytes stayed for too long (more than 1 min) in the vitrification medium before they were stored in liquid nitrogen.
*Compared with the control group p > 0.05.
**Compared with the control group p < 0.05.
DNA fragmentation of the oocytes
The DNA fragmentation in the oocytes was detected by TUNEL technology., which is a qualitative method, not a quantitative one. In other words, we can only tell whether an oocyte had fragmented DNA or not, not how much DNA is fragmented. We considered the oocytes with positive fluorescence as oocytes with DNA fragmentation (Fig. 2). The percentages of oocytes with fragmented DNA in the three study subgroups are higher than that in the control group, however, the difference is not statistically significant for any of the three subgroups (p > 0.05). Within the three subgroups, the percentage of oocytes with fragmented DNA is similar to each other (see Table 2 for details). When the three subgroups were considered as a whole study group, the difference between it and the control group is not significant either.

Figure 2 The DNA fragmentation of the oocytes. The oocyte in (a) and (b) is the same one stained with DAPI and TUNEL, respectively. It is from the positive control group. (a) Shows the figuration and position of the chromosomes. (b) Shows that the oocyte has fragmented DNA in the position of the chromosomes. The oocytes in (c) and (d) are also the same ones from the negative control group. They were stained with DAPI and TUNEL, respectively. The oocytes in (e) and (f) are from the study group. This picture shows that one of the three oocytes has fragmented DNA (as pointed out by the arrow).
Table 2 The oocytes without fragmented DNA in different groups.

*Compared with the control group p > 0.05.
**Compared with the control group p < 0.05.
Fertilization and developmental potential of the oocytes
There were 432 oocytes that were inseminated. A total of 351 of them were fertilized and the fertilization rate was 81.3%. Out of the 351 fertilized oocytes, 267 developed into the blastocyst stage on day 5, and the blastocyst formation rate was 76.1% (267/351). In terms of both fertilization rate and blastocyst formation rate, the difference between the control group and the study group was not statistically significant (Table 3).
Table 3 The fertilization rates and blastocyst formation rates in different groups.

*Compared with the control group p > 0.05.
**Compared with the control group p < 0.05.
Oocytes with improper vitrification manipulation
The improper manipulations in this study included two situations: loading oocytes with too much media on the cryoloop and leaving oocytes in the vitrification medium for more than 60 s.
The survival rates of the oocytes with improper vitrification manipulations were significantly lower than those of the control group (Table 4). Injuries to the spindles, the chromosomes and DNA were all significantly more severe than the control group (Table 1 and Table 2). Only a few oocytes in these two groups got fertilized, yet none developed into blastocyst stage (Table 3).
Table 4 The survival rates of the oocytes with improper manipulations.

*Compared with the control group p < 0.05.
Discussion
Since the first cryopreserved-oocyte baby was born in 1986, only about 200 babies have been born using this technique, Among them, only a small portion used vitrification technique (Oktay et al., Reference Oktay, Cil and Bang2006).
There are two key steps in oocyte cryopreservation. One is picking the optimal developmental stages of oocytes for cryopreservation. The other one is selecting the suitable cryoprotectants, including the medium type and the concentration of reagents, as well as the exposure time of oocytes in the cryoprotectant. Making the right choices in these two steps is critical for thorough permeation of cryoprotectants in the oocytes. Meanwhile, the exposure time of oocytes in cryoprotectant medium should not be too long to avoid damage to oocytes due to chemical toxicity and osmotic shock.
Some studies showed that cryopreservation of immature oocytes got better results than mature oocytes (Frydman et al., Reference Frydman, Selva, Bergere, Auroux and Maro1997). The theory behind this practice is that mature oocytes have a special structure called spindle, which is susceptible to disruption at low temperatures (Chen et al., Reference Chen, Wang, Tsai, Hsieh, Wang and Soong2004; Stachecki et al., Reference Stachecki, Munné and Cohen2004; Bianchi et al., Reference Bianchi, Coticchio, Fava, Flamigni and Borini2005; Coticchio et al., Reference Coticchio, Santis, Rossi, Borini, Albertini, Scaravelli, Alecci, Bianchi, Nottola and Cecconi2006). However, in clinical practice, cryopreservation of immature oocytes is restricted by the technique of in vitro maturation (IVM), which is needed to transform these cryopreserved premature oocytes into fully mature ones when they are needed. To date, no satisfactory IVM methods has been developed yet (Suikkari et al., Reference Suikkari and Söderström-Anttila2007), so we chose to cryopreserve mature oocytes and hoped to obtain better results by modifying some critical steps of the vitrification protocol,
Different cryoprotectants have different permeation and vitrification abilities. To get an ideal vitrification state, many researchers have attempted to improve the cryopreservation vehicles, accelerate the cooling speed, and search for the optimal cryoprotectant combination. However, the existing vitrification protocols differ from each other greatly. Our vitrification protocol was modified from the one used for human blastocysts in our hospital (Sun et al., Reference Sun, He, Yu, Deng and Liu2005). We combined ME2SO and EG, each at 15%, which is lower than the 20% concentration reported in previous study (Cai et al., Reference Cai, Chen, Lian, Zheng and Peng2005). Ficoll 400 and sucrose were added to the vitrification medium as well. Our new protocol used cryoloop as the vehicle, making the vitrification technique easy to master.
Formed with microtubules, the spindles are dynamic structures facilitating chromosomes separation during mitosis or meiosis. They can help chromosomes separate evenly, thereby avoid the formation of aneuploids (Almeida et al., Reference Almeida and Bolton1995). Some studies showed that the injured spindles could recover by themselves after the oocytes were cultured in vitro for several hours (Stachecki et al., Reference Stachecki, Munné and Cohen2004). Using the non-invasive methods (e.g. spindle view imaging system), Rienzi et al. (Reference Rienzi, Martinez, Ubaldi, minasi, Iacobelli, Tesarik and Greco2004) observed that the spindles of oocytes cryopreserved by slow cooling could also self-recover. In our vitrification protocol, we fixed and stained spindles and chromosomes with the immunofluorescence method, and then checked the spindle and chromosome configurations with the fluorescent confocal scanning laser microscope. Compared with the control group, the study group was not statistically different in terms of spindle injury and chromosome distributions. There was also no significant difference between different subgroups, which were divided based on how long the oocytes were incubated in vitro after warmed up. These results are consistent with a report by Larman et al. (Reference Larman, Minasi, Rienzi and Gardner2007). Their study showed that a vitrification protocol at 37 °C had little effect on meiotic spindles, which contradicted the conventional view that cryopreservation would damage oocytes' spindles (Chen et al., Reference Chen, Wang, Tsai, Hsieh, Wang and Soong2004; Stachecki et al., Reference Stachecki, Munné and Cohen2004; Bianchi et al., Reference Bianchi, Coticchio, Fava, Flamigni and Borini2005; Coticchio et al., Reference Coticchio, Santis, Rossi, Borini, Albertini, Scaravelli, Alecci, Bianchi, Nottola and Cecconi2006).
Some studies suggested that DNA molecules in oocytes can be damaged by cryopreservation (Men et al., Reference Men, Monson, Parrish and Rutledge2003). A research (Men et al., Reference Men, Monson, Parrish and Rutledge2003) of bovine oocytes cryopreservation revealed that some of the oocytes that survived morphologically had fragmented DNA – a severe form of DNA injury. DNA injuries would affect the oocytes' potential to be fertilized and develop into blastocysts. In some cases, even the development of fetus could be impaired. Our study showed that vitrification did not cause significant injuries to the oocytes' DNA, and the number of oocytes with DNA fragmentation didn't increase with prolonged post-warming incubation time. However, we did not have conditions with more than 2 h of post-warm-up incubation, and could not tell whether there would be more DNA fragmentation in those cases. The reason for omitting a longer incubation time (>2 h) is that unfertilized oocytes will go through apoptosis when left in an in vitro culture for too long. If we had time points of more than 2 h, we would not be able to tell whether the cell death is caused by in vitro culture or cryopreservation.
Previous research (Men et al., Reference Men, Monson, Parrish and Rutledge2003) indicated that cryopreservation would damage the DNA of oocytes, yet we got quite different results using this new method. Although the mechanism of DNA fragmentation caused by cryopreservation is not fully understood, there are two possible reasons for this contradiction. One is that previous research mainly studied bovine oocytes and we studied mouse oocytes, and the difference between species may lead to different results. The other explanation has something to do with the vitrification vehicle. We used a cryoloop, while other investigators used OPS or ministraws. Using the latter vehicles left much more medium around the oocytes when vitrified than using the cryo loop did, thus would decrease the cooling rate, and cause damages to the oocytes. We modified the vitrification protocol and got promising results with mouse oocytes, indicating that injuries to oocytes by cryopreservation could be avoided by optimizing the cryopreservation protocol.
So far, the highest survival rate of cryopreserved mouse oocyte in previous studies is 99.3% (Lane et al., Reference Lane and Gardner2001) with the ‘ED16 + 10 mg/ml Ficoll + 0.65 M sucrose’ vitrification protocol. With our protocol, the survival rate of oocytes is 98.2%, which is very close to their result. For oocytes, surviving the cryopreservation is just the first step, the more important step is getting fertilized. Many studies demonstrate that due to improper release of cortical granules to the perivitelline space, cryopreservation can harden the oocytes' zona pellucida, leading to failed fertilization (Ghetler et al., Reference Ghetler, Skutelsky, Ben Nun, Ben Dor, Amihai and Shalgi2006). Most researchers believe that the improper release of cortical granules is caused by the chemical toxicity and osmotic shock of cryoprotectants (Larman et al., Reference Larman, Sheehan and Gardner2006). In order to solve this problem, some researchers drilled in hardened zona pellucida, while others added bovine serum albumin, which is the main component of fetal bovine serum (FBS), in the vitrification media (Carroll et al., Reference Carroll, Wood and Whittingham1993). However, adding FBS to the medium makes the procedure quite different from what happens naturally. Besides FBS can not be used on human oocytes since it may transmit diseases. This problem of low fertilization rate of thawed human oocytes remained unsolved until the introduction of ICSI in 1992 (Palermo et al., Reference Palermo, Joris, Devroey and Van Steirteghem1992; Kazem et al., Reference Kazem, Thompson, Srikantharajah, Laing, Hamilton and Templeton1995; Porcu et al., Reference Porcu, Fabbri, Seracchioli, Ciotti, Magrini and Flamigni1997). In our study, the zona pellucida of oocytes did not get any treatment, and all cryopreserved oocytes were directly inseminated in vitro. The fertilization rate was 81.3%. This result indicated that the hardening of oocytes' zona pellucida by cryopreservation, which may result in fertilization failure, could be avoided by adopting a proper vitrification protocol.
Although our protocol generated good results when followed correctly, improper manipulations can lead to severe damage to oocytes. For example, too much vitrification media on the cryoloop and exposing oocytes in the vitrification media for a long time are both harmful to oocytes.
In conclusion, the cryoloop vitrification protocol was used to vitrify mature mouse oocytes in our study. This protocol achieved a high survival rate of oocytes with little damage to the spindles/chromosomes and DNA. It also preserved the developmental potential of oocytes, which led to satisfactory developmental results. However, mouse oocytes are quite different from human oocytes in many characteristics, and more research is needed to examine this protocol in human. Specifically, the vitrification and warm-up procedures at room temperature should be thoroughly studied and confirmed to be safe before using this protocol in clinics.
Acknowledgements
This study was supported by grants 2008 BAI 57B02 from the Ministry of Science and Technology of China. We would like to thank Sharan Athwal for her editing and Suggestions.






