Introduction
Although antibiotics and vaccines are widely used, infectious diseases caused by intracellular pathogens still result in great economic loss. Previous studies have demonstrated that natural resistance-associated macrophage protein 1 (Nramp1) controls innate resistance and susceptibility to intracellular pathogens such as Mycobacterium, Leishmania, Salmonella, and Brucella (Govoni et al., Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999; Barthel et al., Reference Barthel, Feng, Piedrahita, McMurray, Templeton and Adams2001). The Nramp1 gene was first cloned from mouse by the positional cloning method (Vidal et al., Reference Vidal, Malo, Vogan, Skamene and Gros1993). cDNA, genomic DNA and trafficking of the bovine Nramp1 gene have been characterized subsequently (Feng et al., Reference Feng, Li, Hashad, Schurr, Gros, Adams and Templeton1996; Cheng et al., Reference Cheng, Deng, Meng, Lai and Wang2011; Cheng & Wang, Reference Cheng and Wang2012). The NRAMP1 protein is a hydrophobic integral membrane protein that consists of 12 transmembrane domains (TMD) (Barton et al., Reference Barton, White, Roach and Blackwell1994). NRAMP1 is expressed in granules of polymorphonuclear leukocytes (Cellier et al., Reference Cellier, Shustik, Dalton, Rich, Hu, Malo, Schurr and Gros1997; Canonne-Hergaux et al., Reference Canonne-Hergaux, Calafat, Richer, Cellier, Grinstein, Borregaard and Gros2002), late endosomes and lysosomes in macrophages (Gruenheid et al., Reference Gruenheid, Pinner, Desjardins and Gros1997; Searle et al., Reference Searle, Bright, Roach, Atkinson, Barton, Meloen and Blackwell1998) and in dendritic cells (Stober et al., Reference Stober, White, Popoff and Blackwell2007). NRAMP1 can influence the replication of intracellular pathogens by mediating the activity of macrophages during the early stages of infection (Blackwell et al., Reference Blackwell, Barton, White, Roach, Shaw, Whitehead, Mock, Searle, Williams and Baker1994), such as by depleting phagolysosomal iron, which is needed for pathogens survival (Forbes & Gros, Reference Forbes and Gros2001). NRAMP1 also stimulates the production of nitric oxide, a potent antimicrobial effector, by decreasing intracellular iron availability (Fritsche et al., Reference Fritsche, Nairz, Theurl, Mair, Bellmann-Weiler, Barton and Weiss2007).
A naturally occurring glycine to aspartic acid substitution at position 169 (G169D) within the mouse Nramp1 gene results in NRAMP1 loosing its ability to inhibit the replication of intracellular pathogens such as Salmonella and Brucella (Malo et al., Reference Malo, Vogan, Vidal, Hu, Cellier, Schurr, Fuks, Bumstead, Morgan and Gros1994; Vidal et al., Reference Vidal, Tremblay, Govoni, Gauthier, Sebastiani, Malo, Skamene, Olivier, Jothy and Gros1995). In addition, the mouse macrophage cell line RAW264.7, containing a defective NRAMP1 (G169D), has been used widely to determine the Nramp1 function in vitro (Govoni et al., Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999; Barthel et al., Reference Barthel, Feng, Piedrahita, McMurray, Templeton and Adams2001). Previous reports have shown that recombinant mouse NRAMP1 protein in RAW264.7 cells abrogated the replication of Salmonella typhimurium (Govoni et al., Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999).
Expression of tissue-specific therapeutic genes in transgenic animals is an alternative approach to conventional strategies to avoid the deleterious side effects in the whole body. It has been reported that synthetic promoter (SP) was capable of driving the targeted gene expression especially in macrophages, which provided an ideal tool for driving the Nramp1 gene to control intracellular bacterial infections in macrophages (He et al., Reference He, Qiang, Ma, Valente, Quinones, Wang, Reddick, Xiao, Ahuja, Clark, Freeman and Li2006; Wen et al., Reference Wen, Guo, Yu, Cen, Liu, Chen, Zhang and Tu2008, Reference Wen, Yu, Xia, Cen, Liu and Tu2009). Genetic manipulation combined with somatic cell nuclear transfer (SCNT) is a promising strategy to generate transgenic cloned cattle with a heightened resistance to infections by intracellular pathogens.
The aim of present study was to evaluate the function of bovine Nramp1 in vitro and to produce Nramp1 transgenic cloned embryos. This study may provide a valuable reference for the production of Nramp1 transgenic cattle in the future.
Materials and methods
Nramp1 cDNA cloning and vector construction
Primers NRAMP1-F and NRAMP1-R were designed based on GenBank sequence NM_174652.2 (Table 1). The HA tag sequence was fused with the C terminus of Nramp1 in order to evaluate exogenous Nramp1 expression. To clone the Nramp1 gene, total RNA was extracted from the spleen of Qinchuan cattle using Trizol reagent (Invitrogen, USA). Total RNA was reverse transcribed to cDNA with oligo-dT primers and RevertAid™ reverse transcriptase (Fermentas, Canada). Bovine Nramp1 gene amplified by Triplemaster PCR system (Eppendorf, Germany) was cloned into the pGEM-T-Easy vector (Promega, USA) and sequenced. The positive clone was named pGEM-T-Easy–NRAMP1–HA.
Table 1 Primers used in this study

aKozak and HA tag sequences are underlined in NRAMP-F and NRAMP1-R, respectively. SacII and BamHI sites are shown in bold in NRAMP-F and NRAMP1-R, respectively.
To construct expression vectors, pEGFP-N1 (Invitrogen, USA) and the macrophage-specific expression vector pSP-EGFP (a gift from Dr Wen Yangan, Chongqing Medical University, China) were digested with SacII and BamHI, and the Nramp1 gene from pGEM-T-Easy–NRAMP1–HA was subcloned into plasmids to construct pCMV–NRAMP1–HA–GFP and pSP–NRAMP1–HA–GFP, respectively. Plasmid pCMV–NRAMP1–HA was constructed by deleting the GFP coding sequence and validated by PstI digestion. The pSP–NRAMP1–HA vector without GFP was constructed and validated by XhoI and HpaI enzyme digestion.
Cell culture and transfection
RAW264.7 cells, an immortalized mouse macrophage cell line containing a non-functional NRAMP1 gene, were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) high-glucose formulation supplemented with 10% fetal bovine serum (HyClone, USA). In order to evaluate the expression of Nramp1 constructs, RAW264.7 cells plated on 24-well plates were transfected with pSP–NRAMP1–HA and pCMV–NRAMP1–HA, respectively, using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. Twenty-four hours after transfection, total RNA was extracted and subjected to RT-PCR assay using Nramp1-specific primers F1 and R1 (Table 1 and Fig. 1A). Primer R1 was within the HA tag sequence. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control.
Figure 1 Nramp1 gene cloning and expression in macrophage RAW264.7 cells. (A) Schematic representation of pCMV–NRAMP1–HA (pCMV-N) plasmid driven by cytomegalovirus (CMV) promoter and pSP–NRAMP1–HA (pSP-N) plasmid driven by macrophage-specific synthetic promoter (SP). The Nramp1 coding sequence was fused with an HA tag. Primers F1 and R1 were designed to determine exogenous Nramp1 expression. (B) RT-PCR analysis of Nramp1 expression in RAW264.7 cells. Lane 1, Cells with pSP–NRAMP1–HA; Lane 2, Cells with pCMV–NRAMP1–HA; Lane 3, RAW264.7 cells. GAPDH was used as internal control. (C) RAW264.7 cells were transfected with pSP–NRAMP1–HA (1 and 2) and pCMV–NRAMP1–HA (3 and 4), respectively. The expression of Nramp1 was determined by anti-HA antibody followed by secondary antibody conjugated with TRITC. (D) Co-localization of NRAMP1 and Lamp1 in CHO cells. pCMV–NRAMP1–HA was co-transfected using Lamp1-RFP into CHO cells. (1) Immunofluorescence due to NRAMP1 (green) was detected with anti-HA antibody and FITC-conjugated anti-mouse IgG; (2) Lamp1-RFP expression (red); (3) DAPI staining and merged image.
To determine the subcellular localization of NRAMP1, the Lamp1-RFP plasmid, which was the lysosome marker and purchased from Addgene, was co-transfected with pCMV–NRAMP1–HA into Chinese hamster ovary (CHO) cells using the Lipofectamine 2000. To perform the immunofluorescence assay, cells at 36 h after transfection were washed twice with phosphate-buffered saline (PBS; Gibco, USA) and fixed in 4% paraformaldehyde for 20 min. An immunofluorescence assay was performed using mouse monoclonal anti-HA antibody (GenScript, USA, 1:500) as described previously (Lam-Yuk-Tseung et al., Reference Lam-Yuk-Tseung, Picard and Gros2006). Cells were then incubated in 100 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) for 5 min and visualized under a fluorescence microscope (Nikon, Japan).
In order to determine the antibacterial effect of bovine Nramp1, RAW264.7 cells were seeded onto 6-cm plates 1 day before transfection. Plasmids pCMV–NRAMP1–HA and pSP–NRAMP1–HA were transfected into cells using the Lipofectamine 2000 method. At 48 h after transfection, medium containing 400 μg/ml G418 (Gibco-BRL, USA) was added into the plates. After 10–12 days of incubation, the individual G418-resistant clones were picked and expanded. Stably transfected clones were confirmed by semi-quantitative RT-PCR and F1 and R1 primers.
To obtain reliable transgenic donor cells for SCNT, calf fibroblasts (CF) were generated from the ear skin of newborn female Qinchuan calf by the tissue culture method described previously (Lipiński et al., Reference Lipiński, Duszewska, Zeyland, Mały, Gawron, Rynkowska, Reklewski and Słomski2007). CF cells were transfected with pSP–NRAMP1–HA using Lipofectamine 2000. After 48 h of transfection, cells were transferred into medium that contained 800 μg/ml of G418 and cultured for 2 weeks. Drug-resistant clones were subcultured and confirmed by RT-PCR. Cytogenetic analysis and the proliferation potential of transgenic fibroblasts were assessed based on a previous report (He et al., Reference He, Wu, He, Liu, He and Zhang2009).
Antibacterial assay
Assays for bacteria strains Salmonella abortusovis and Brucella abortus were performed as described previously (Barthel et al., Reference Barthel, Feng, Piedrahita, McMurray, Templeton and Adams2001). Briefly, RAW264.7 macrophages (as the control) and Nramp1 transfected RAW264.7 (RAW264/NRAMP1) cells were grown to 80% confluency. Fifty thousand cells were seeded onto 12-well plates, one plate was used to determine the number of bacteria at the starting point, and the other plates were used to determine the number of bacteria at 24 h after infection. Macrophages from both control and RAW264/NRAMP1 cells were challenged with 5 × 106 bacteria for 2 h, respectively, to allow for phagocytosis. To inhibit the growth of extracellular bacteria, 25 μg/ml of G418 was added for 1 h, and cells were washed with PBS three times. Control cells were collected to count the number of colony-forming units at the starting point (CFU0), and experimental cells were incubated at 37°C for 24 h. Macrophages were lysed osmotically with 0.4 ml 0.01% bovine serum albumin (BSA), and cell lysates were serially diluted and sprayed onto agar plates for the CFU count (CFU24). Replication ability of the bacteria was represented by the ratio of CFU24 versus CFU0.
Production of transgenic cloned embryos
To produce SCNT embryos, CFs and an Nramp1 transgenic calf fibroblast clone (SP-CF2) with a normal karyotype were used as donor cells, which were grown in the 6-well plate until 80% confluency, and were then trypsinized. High quality bovine oocytes isolated from ovaries and used to perform SCNT were matured in TCM199 (Gibco, USA) medium supplemented with 2.5 μg/ml sodium pyruvate, 50 μl/ml insulin–transferrin–selenium (ITS; Gibco, USA), 0.1 IU/ml human menopausal gonadotropin (HMG; Ningbo animal hormone factory, China), 1 μg/ml estradiol (Ningbo animal hormone factory, China) and 10 ng/ml epidermal growth factor (EGF; Millipore, USA) for 22 h. One donor cell was inserted into the perivitelline space of an enucleated oocyte. Electrofusion was carried out with two direct current pulses of 140 V/mm for 80 μs, with a 1 s interval. After electrofusion, the reconstructed oocytes were cultured at 38°C in a 5% CO2 in air atmosphere for 6–7 days. DNA isolated from a single reconstructed embryo at the morula or blastocyst stages was analyzed by PCR using primers F2 and R2 as reported previously (Melo et al., Reference Melo, Sousa, Iguma, Franco, Rech and Rumpf2005; Zhang et al., Reference Zhang, Wan, Wang, Xu, Pang, Meng, Wang, Zhong and Wang2010). The transgenic hatched blastocysts were incubated in 100 ng/ml DAPI for 5 min, washed three times in PBS, mounted on slides and squashed under a coverslip. Blastocysts were visualized with a fluorescence microscope (Nikon, Japan).
Embryo transfer and pregnancy diagnosis
Animal experiments were performed under animal welfare standards issued and approved by Animal Research Committee of Northwest A&F University (No. 001004150A). To perform embryo transplantation, 86 blastocysts were produced by SCNT using SP-CF2 transgenic fibroblasts. Two reconstructed Nramp1 transgenic blastocysts were transferred to each recipient bovine (n = 43) as described previously (Jang et al., Reference Jang, Bhuiyan, Jeon, Ko, Park, Kim, Kim, Kang, Lee and Hwang2006). At 60 days after transplantation, pregnancy was determined by rectal palpation. Thereafter, the different pregnancy phases were monitored by rectal palpation and ultrasound every 2 months.
Detection of Nramp1 transgene in the cloned fetus
To evaluate the integration of Nramp1 transgene from the cloned fetus, genomic DNA was isolated and PCR was performed using primers F1 and R1. In order to confirm the transgene insertion, primers F2 and R2 (Table 1 and Fig. 5B) were designed and used to perform PCR.
Statistical analysis
SCNT experiments were repeated at least three times. Data on development of the SCNT embryos were analyzed using the chi-squared test (SAS, USA). Significant difference was confirmed when the P-value was less than 0.05.
Results
Bovine Nramp1 cDNA cloning and expression in macrophages
The HA tag sequence was fused with the C terminus of cloned Nramp1 cDNA in order to distinguish the exogenous and endogenous Nramp1 gene (Table 1 and Fig. 1A). The cDNA sequence was amplified from the spleen of Qinchuan cattle. When compared with the Nramp1 sequence published in GenBank (NM_174652.2), the cloned sequence displayed 99% identity except for two nucleotide variations. One variation (A to G) was located at the 145th position of the coding region that resulted in a threonine to alanine residue change. The second variation (A to C) located at the 157th position of the coding region did not result in an amino acid change. The Nramp1 gene was subcloned into pSP-EGFP and pEGFP-N1 plasmids, respectively, which were confirmed by DNA sequencing (Fig. 1A).
RT-PCR and the immunofluorescence assay were used to evaluate the expression of exogenous Nramp1 gene in macrophages. Exogenous Nramp1 was identified in the transfected macrophages, but was absent from the untransfected macrophages (Fig. 1B). To verify the expression of Nramp1 protein, immunofluorescence analysis using the anti-HA antibody was conducted and the recombinant protein NRAMP1 fused with HA tag was detected in macrophages (Fig. 1C). The lysosomal localization of NRAMP1 protein was confirmed by co-transfecting the NRAMP1 construct and the LAMP1 (lysosome associated membrane protein 1, lysosome marker) construct into CHO cells and determining protein expression by immunofluorescence assay (Fig. 1D). The result was consistent with that of previous reports (Lam-Yuk-Tseung et al., Reference Lam-Yuk-Tseung, Picard and Gros2006; Cheng & Wang, Reference Cheng and Wang2012).
NRAMP1 inhibited the growth of Salmonella and Brucella
Overexpression of mouse NRAMP1 protein has been shown to inhibit the replication of intracellular pathogens such as Salmonella and Brucella in the macrophage (Govoni et al., Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999). In order to test the function of bovine NRAMP1, two expression plasmids, pCMV–NRAMP1–HA driven by cytomegalovirus (CMV) promoter and pSP–NRAMP1–HA driven by macrophage-specific SP, were constructed and transfected into RAW264.7 macrophages. A representative clone that was stably transfected with pCMV–NRAMP1–HA construct is shown in Fig. 2A. RT-PCR analysis showed the expression of exogenous Nramp1 in stably transfected RAW264.7 cell lines from both CMV–NRAMP1 and SP–NRAMP1 constructs (Fig. 2B). Anti-Salmonella abortusovis and anti-Brucella abortus assays were performed based on these stably transfected cell lines. In comparison with untransfected RAW264.7 macrophages (control), both pCMV–NRAMP1–HA and pSP–NRAMP1–HA transfectants significantly inhibited the growth of Salmonella abortusovis and Brucella abortus (Fig. 2C). Of note, the Nramp1 macrophage transfectants with a SP promoter displayed similar levels of inhibition as did those with the CMV promoter, further confirming that the SP promoter could be utilized to efficiently direct Nramp1 expression in macrophages and to produce Nramp1 transgenic bovines (Fig. 2C).
Figure 2 Antimicrobial assay. (A) Morphology of RAW264.7 macrophage clone that was stably transfected with exogenous Nramp1 cDNA. (B) RT-PCR analysis of stably transfected RAW264.7 clones. Lane 1, RAW264.7 with pCMV–NRAMP1–HA; Lane 2, RAW264.7 with pSP–NRAMP1–HA; Lane 3, RAW264.7 cell as the negative control. GAPDH was used as internal control. (C) Antimicrobial testing Salmonella abortusovis and Brucella abortus survival after three different treatments. Control (Con), the macrophage RAW264.7 cells infected with Salmonella and Brucella strains; pCMV-N, RAW264.7 cells with pCMV–NRAMP1–HA plasmid; pSP-N, RAW264.7 cells with pSP–NRAMP1–HA plasmid. The results are presented as a mean ± SD (n = 3) and as the percentage of inhibition versus control cells. **P < 0.01.
Reconstruction of transgenic embryos
CFs were isolated from the ear skin of newborn female Qinchuan calf by the tissue culture method described previously (Lipiński et al., Reference Lipiński, Duszewska, Zeyland, Mały, Gawron, Rynkowska, Reklewski and Słomski2007) (Fig. 3A). To characterize the stably transfected fibroblasts, 16 clones were picked and cultured in medium that contained G418. Five G418-resistant clones that contained the transgene Nramp1 were identified (Fig. 3B). One stably transfected clone SP-CF2 showed a normal karyotype containing 30 pairs of chromosomes (2n = 58 + XX; Fig. 3D). The proliferation potential of SP-CF2 was measured compared with control CF cells, showing that the doubling time of SP-CF2 is 37.9 h versus 34.8 h for CFs (Fig. 3C).
Figure 3 Characterization of stably transfected cell lines. (A) Morphology of isolated newborn calf fibroblasts (CF). (B) Five stably transfected fibroblast clones harbouring pSP–NRAMP1–HA plasmid were analyzed by PCR to detect the insertion of exogenous Nramp1. Lane 1, newborn calf fibroblasts; Lanes 2–6, stably transfected clones. Clone SP-CF2 shown in Lane 3; Lane 7, pSP–NRAMP1–HA plasmid is the positive control; M, DNA marker. (C) Growth curve of clone SP-CF2 and the calf fibroblasts (CF). (D) Karyotype of clone SP-CF2.
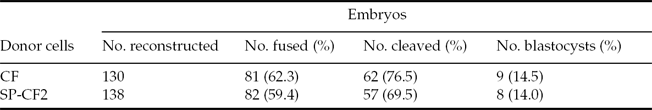
The results of 138 cloned embryos derived from the SP-CF2 group and 130 cloned embryos derived from the CF group showed that the fusion rate between the SP-CF2 group and CF group did not differ significantly (59.4% versus 62.3%). Moreover, the blastocyst formation rate of SP-CF2 embryos (14.0%) was similar to that of CF embryos (14.5%, P > 0.05) (Table 2). The reconstructed embryos could be developed in vitro to maturate into the hatched blastocyst stage (Fig. 4A). PCR results further confirmed that the reconstructed embryos retained the Nramp1 transgene (Fig. 4B).
Figure 4 Reconstruction of bovine embryos. SP-CF2 cells and calf fibroblasts (CF) were used as donor cells to perform somatic cell nuclear transfer (SCNT). (A) A hatched blastocyst was derived by SCNT of SP-CF2 cells. (B) PCR analysis of Nramp1 gene integration in reconstructed embryos with primers F1 and R1. Lane 1, SP-CF2 cells; Lane 2, embryos derived from SCNT of CF; Lanes 3–6, embryos derived from SCNT of SP-CF2; Lane 7, plasmid DNA of pSP–NRAMP1–HA; M: DNA marker.
Table 2 In vitro maturation of bovine embryos reconstructed with calf fibroblast (CF) or SP-CF2 cells
Transplantation of embryos reconstructed with SP-CF2 cells and characterization of transgenic bovine fetus
The transgenic embryos in the blastocyst stage were transplanted into 43 Qinchuan recipient cattle. At 60 days after transplantation, pregnancy tests were performed by rectal palpation and the recording of a non-return estrus, showing that 14 recipients were pregnant. At 90 days after transplantation, rectal palpation and ultrasonography were then applied to diagnose the 14 pregnant recipients. At this time, three recipients were confirmed to have remained pregnant, while the pregnancy of the remaining animals (11) had been terminated due to the higher percentage of first and second trimester embryonic loss or absorption, which has often been reported following the SCNT cloning (Kato et al., Reference Kato, Tani and Tsunoda2000). Within the first 6½ months, miscarriages occurred in three pregnant cattle at the different developmental stages because of the abnormal development of fetus or placenta (Farin et al., Reference Farin, Piedrahita and Farin2006). One aborted fetus at 6½ months pregnancy is shown in Fig. 5A. To confirm the integration of the transgene, we designed one specific set of F2 and R2 primers, which were used to differentiate the exogenous and endogenous Nramp1 gene simultaneously (Fig. 5B). The PCR results of specimens from aborted fetal brain, skin and umbilical cord tissue showed that the aborted fetus had retained the Nramp1 transgene (Fig. 5C,D). Due to the decomposition of aborted fetus in recipient bovine uterus over several days, DNA isolated from tissue of muscle, kidney, liver and spleen were degraded heavily and unable to be used for PCR.
Figure 5 Characterization of transgenic bovine fetus. (A) An aborted fetus from the transplantation of embryos reconstructed with SP-CF2 cells. (B) Schematic representation of the exon 14 to 15 region of bovine Nramp1 gene and the location of primers. The PCR products of 318 bp and 182 bp fragments are from endogenous and exogenous Nramp1 gene, respectively. (C, D) PCR analysis of transgene in the transplant fetal tissues with primers F2/R2 (C) and primers F1/R1 (D). Lane 1, calf fibroblasts; Lane 2–4, specimens of brain, skin and umbilical cord from aborted fetus tissues; Lane 5, SP-CF2 cells; Lane 6, pSP–NRAMP1–HA plasmid DNA as positive control; Lane 7, H2O as negative control. M, DNA marker.
Discussion
Intracellular pathogens are challenges in the treatment and control of refractory intracellular infectious diseases as they are ingested and protected in macrophages, thus resistant to the host humoral immunity. There are few effective methods for efficiently controlling intracellular pathogens, which cause great loss in livestock production. Previous reports have shown that Nramp1 is one candidate gene for controlling the replication of intracellular pathogens (Govoni et al., Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999; Barthel et al., Reference Barthel, Feng, Piedrahita, McMurray, Templeton and Adams2001). Here we determined the function of bovine Nramp1 in macrophages and illustrate the production of a Nramp1 transgenic cloned embryos in vitro.
Although the Nramp1 gene is conserved in both prokaryotes and eukaryotes (Cellier et al., Reference Cellier, Belouchi and Gros1996), there are still polymorphisms among species and even among different breeds. Nramp1 cDNA was cloned from the spleen of Qinchuan cattle in this study; sequencing results showed that there were two nucleotide differences within the amplified Nramp1 sequence when compared with the bovine Nramp1 sequence from GenBank. This finding was confirmed by two further rounds of Nramp1 gene cloning, suggesting that these differences might be due to polymorphism in the Nramp1 gene between Qinchuan cattle and other bovine species.
In this study the macrophage cell line RAW264.7 was used to evaluate the functionality of macrophage-specific SP and to examine recombinant bovine Nramp1 gene expression in vitro. The RAW264.7 cell line contains non-functional NRAMP1 with a single glycine to aspartic acid substitution at position 169 (G169D) (Govoni et al., Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999). In addition, overexpression of NRAMP1Gly169 protein in RAW macrophages (NRAMP1Asp169) restores these cells to inhibit the replication of Salmonella typhimurium (Govoni et al., Reference Govoni, Gauthier, Billia, Iscove and Gros1997, Reference Govoni, Canonne-Hergaux, Pfeifer, Marcus, Mills, Hackam, Grinstein, Malo, Finlay and Gros1999; Barthel et al., Reference Barthel, Feng, Piedrahita, McMurray, Templeton and Adams2001). Although bovine macrophage cell lines have already been established (Matsumoto et al., Reference Matsumoto, Tamura, Denhardt, Obinata and Noda1995; Stabel & Stabel, Reference Stabel and Stabel1995), previous reports have shown that the RAW264.7 cell line containing a defective NRAMP1 is an optimal cell line to investigate Nramp1 function in vitro. Therefore, in this study, we used the mouse macrophage cell line RAW264.7 to determine Nramp1 function in vitro.
Both RT-PCR and immunofluorescence assay revealed that the Nramp1 gene could be expressed in the macrophage. Furthermore, as there are two nucleotide differences in the coding sequence of the bovine Nramp1 gene compared with the published sequence in GenBank, it is important to determine whether the cloned Nramp1 gene could inhibit the replication of intracellular pathogens in the macrophage. In our study, NRAMP1–HA RAW264.7 transfectants showed strong inhibition of replication of both Salmonella abortusovis and Brucella abortus in comparison with the untransfected macrophages. This observation confirms a previous report (Barthel et al., Reference Barthel, Feng, Piedrahita, McMurray, Templeton and Adams2001). Moreover, macrophage-specific SP exhibited similar levels of antibacterial activity as did the CMV promoter in macrophages, which was consistent with previous reports (He et al., Reference He, Qiang, Ma, Valente, Quinones, Wang, Reddick, Xiao, Ahuja, Clark, Freeman and Li2006; Wen et al., Reference Wen, Guo, Yu, Cen, Liu, Chen, Zhang and Tu2008). This result suggested that the Nramp1 gene under SP control could be expressed in macrophage in vivo.
The genetic background of donor cells used for SCNT is a very important factor that may affect the efficiency of producing transgenic animals and transgene expression in cloned livestock (Powell et al., Reference Powell, Talbot, Wells, Kerr, Pursel and Wall2004; Campbell et al., Reference Campbell, Fisher, Chen, Choi, Kelly, Lee and Xhu2007; Zhao et al., Reference Zhao, Lin, Liu, Quan, Wang, Liu, Hua and Zhang2009). For instance, the chromosome number of the donor cells might be altered during long-term culture under G418 selection. In order to obtain high quality Nramp1 transgenic fibroblast cells, we performed G418-resistence selection and single transgenic cell cloning selection was confirmed by PCR analysis. The stably transfected cell line SP-CF2 used for SCNT retained the normal karyotype and was propagated for >20 passages. Reconstructed embryos were able to maturate in vitro and the blastocyst rate was similar to that of the control. This result was consistent with previous reports (Lee et al., Reference Lee, Kumar, Kim, Ock, Jeon, Balasubramanian, Choe and Rho2007; Zhang et al., Reference Zhang, Wan, Wang, Xu, Pang, Meng, Wang, Zhong and Wang2010).
Although live offspring from cattle transgenic for the above cell line have not yet been obtained, the aborted fetus showed transgene insertion in multiple tissues, suggesting that more recipient animals and different cell lines were needed to apply transplantation. Similar observations have reported that recipients were aborted within approximately 90 days (Kato et al., Reference Kato, Tani and Tsunoda2000). Embryonic loss or absorption in the SCNT animal could be triggered by: (1) the inappropriate breeding season, or by allantoic malformation, which occurred during the first trimester (Peterson & Lee, Reference Peterson and Lee2003); or (2) the incomplete epigenetic modification of recombinant embryos that results in the abnormal development of fetus and placenta of cloned animals (Farin et al., Reference Farin, Piedrahita and Farin2006). Because SP is a strong macrophage-specific promoter, we speculate that the Nramp1 transgene under SP promoter control could be expressed in macrophages in cloned cattle. However, the expression of both RNA and protein from transgene in aborted fetus was unable to be detected due to the decay of specimens. In future, the priority is to demonstrate tissue-specific expression of the Nramp1 gene in cloned animals and its biological activity against intracellular pathogens.
Acknowledgements
We thank Mrs Wenlin Yang, Wei Li, Hao Dong and Dr Qingmin Wu for the technical support. We are grateful to Dr Renjith Mathew for critically evaluating the manuscript. This work was supported by the National Transgenic Breeding Project (2009ZX08007–008B), China.