Introduction
Dongxiang wild rice (Oryza rufipogon, DXWR) is a common wild rice that originates in Dongxiang County, Jiangxi Province of China (Li et al., Reference Li, Guo, Zhao, Chen, Chong and Xu2010). DXWR possesses many valuable agronomic traits, such as heterosis, cytoplasmic male sterility, fertility restoration, high yield and resistance to biotic and abiotic stresses (Zhang et al., Reference Zhang, Xu, Mao, Yan, Chen, Wu, Chen, Luo, Xie and Gao2016). Therefore, DXWR is considered as a precious germplasm resource for cultivated rice (Oryza sativa) breeding.
The use of molecular markers can greatly accelerate the breeding process (Ganopoulosa et al., Reference Ganopoulosa, Avramidoua, Fasoula, Diamantidis and Aravanopoulos2010). Simple sequence repeat (SSR) marker is regarded as one of the most efficient and ideal molecular markers (Powell et al., Reference Powell, Machray and Provan1996). SSR marker can be divided into two types, i.e. genomic and genic SSR markers. Compared with genomic SSR marker, genic SSR marker has some intrinsic advantages, such as a wealth of functional annotations, relatively high transferability to closely related species and can be used as anchor markers for comparative mapping and evolutionary studies (Yue et al., Reference Yue, Liu, Zong, Teng and Cai2014). However, the available genic SSR markers for DXWR are still extremely limited due to lack of genomic resource and genome complexity. Therefore, the objectives of this study were to: (1) use transcriptome sequencing to develop a set of genic SSR markers for DXWR and (2) validate the developed genic SSR markers in DXWR and commercial cultivars. The results from this study will greatly enrich the number of molecular markers available in DXWR, which will provide additional tools for studying genetic diversity, linkage mapping, germplasm characterization and molecular marker assisted breeding for this valuable and endangered wild rice germplasm.
Experimental
A total of 16 accessions of rice germplasm were used in this study, including DXWR (Fig. S1, available online) and 15 commercial cultivars (Table S1, available online). The rice materials used in this study were all collected in our laboratory.
The assembled unigene sets were integrated and assembled into non-redundant unigenes using the TIGR Gene Indices Clustering (TGICL) tools (v2.1, http://sourceforge.net/projects/tgicl/files/tgicl%20v2.1/) and CD-HIT program (Pertea et al., Reference Pertea, Huang, Liang, Antonescu, Sultana, Karamycheva, Lee, White, Cheung, Parvizi, Tsai and Quackenbush2003; Li and Godzik, Reference Li and Godzik2006). The parameters of TGICL were set at a similarity of 95% and an overlap length of 40 bp and the sequence identity cut-off for CD-HIT was set to 0.95. Genic SSR loci were identified from unigenes by MISA (http://pgrc.ipk-gatersleben.de/misa), which is based on the Perl program, with criteria of 12, 6, 5, 5, 4 and 4 minimum motifs units for mono-, di-, tri-, tetra-, penta- and hexa-nucleotide repeats, respectively (You et al., Reference You, Liu, Liu, Zheng, Diao, Huang and Hu2015). Primer 3.0 software was used to design PCR primers in the flanking regions of the SSR loci (Untergasser et al., Reference Untergasser, Cutcutache, Koressaar, Ye, Faircloth, Remm and Rozen2012). The primer pair with the highest score was chosen as the best one.
Genomic DNA was extracted according to the CTAB protocol (Porebski et al., Reference Porebski, Bailey and Baum1997). PCR was performed using T100™ Thermal Cycler (Bio-Rad) in a final volume of 15 µl PCR reaction mixture, including 1.5 µl of 10 × PCR buffer (with Mg2+), 2 µl of genomic DNA (50 ng), 0.5 µl of dNTP (10 mM), 1 µl of each primer (10 pmol), 0.5 µl of Taq DNA polymerase (1 unit) and 9.5 µl of ddH2O. PCR amplified fragments were analysed using 3% agarose gels stained with GoodView™.
Discussion
Two cDNA libraries were prepared from the leaf and root section of DXWR under normal condition in our previous study, and they were named as LCK and RCK, respectively. Transcriptome sequencing generated 46,784,432 high-quality reads that were assembled into 82,772 unigenes for LCK, and generated 43,588,908 high-quality reads that were assembled into 75,768 unigenes for RCK (Zhou et al., Reference Zhou, Yang, Cui, Zhang, Luo and Xie2016). In this study, the two transcriptome data were integrated and assembled into a total of 76,258 unigenes.
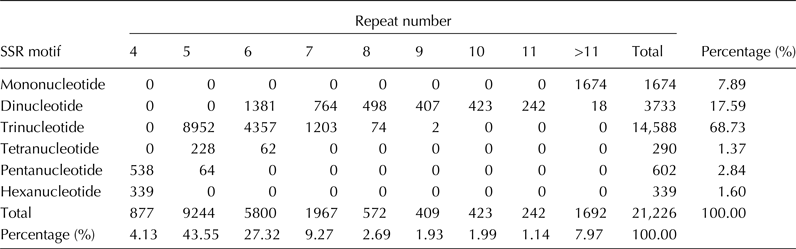
All of the 76,258 unigenes were used to mine SSR loci that were defined as mononucleotide to hexanucleotide SSRs with a minimum of four repetitions for all motifs. A total of 21,226 SSR loci were discovered; and the SSR loci were distributed in 16,311 unigenes, of which 12,524 possessed a single SSR locus, 3787 contained more than one SSR, and 1728 had compound SSRs. Among these SSRs, the trinucleotide (14,588, 68.73%) and dinucleotide (3733, 17.59%) repeat motifs were the most abundant types (Table 1).
Table 1. Frequencies of different SSR repeat motif types
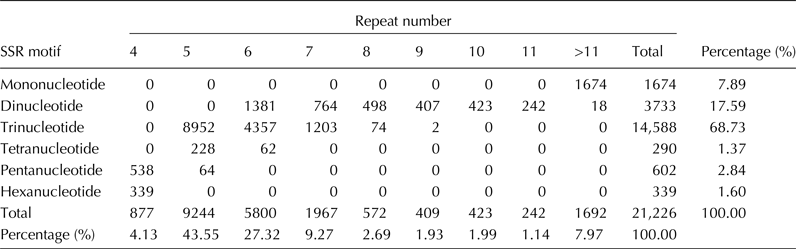
The most abundant dinucleotide repeat was AG/CT with 65.66% of all dinucleotide repeats found in the unigenes (Fig. S2, available online). The CCG/CGG motif (51.27%) was the most abundant trinucleotide repeat type, which supported the view that the abundance of CCG/CGG repeat was a specific feature of monocot genomes (Wang et al., Reference Wang, Li, Luo, Huang, Chen, Fang, Li, Chen and Zhang2011). A total of 3681 primer pairs were successfully designed based on the flanking sequences of each SSR locus, including 7 (0.19%) for mononucleotide repeats, 492 (13.37%) for dinucleotide repeats, 2947 (80.06%) for trinucleotide repeats, 67 (1.82%) for tetranucleotide repeats, 88 (2.39%) for pentanucleotide repeats and 80 (2.17%) for hexanucleotide repeats (Table S2, available online).
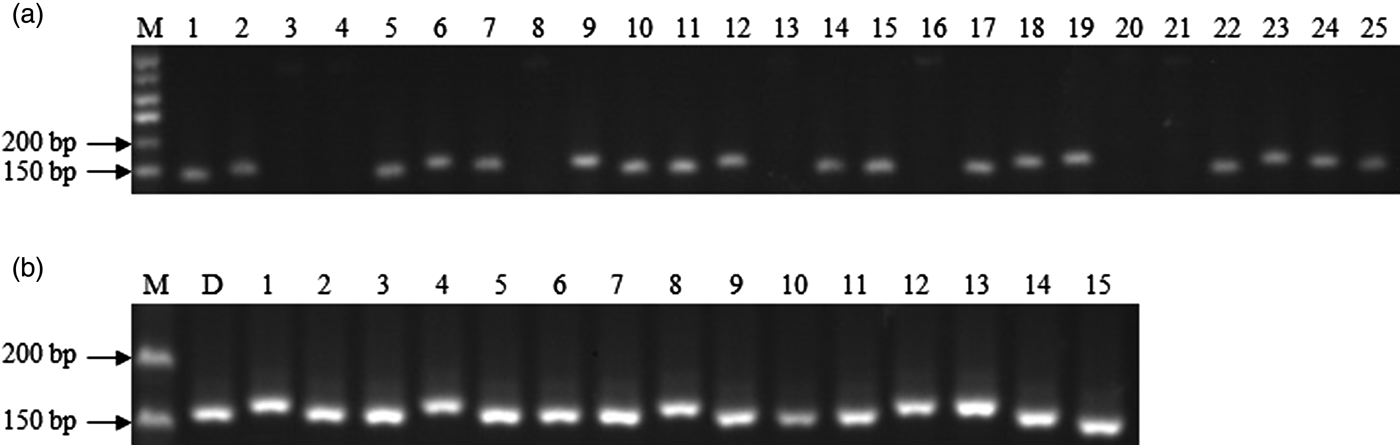
Subsequently, we randomly selected 25 primer pairs for the verification, and 18 (72%) of them were successfully amplified in PCR amplification with genomic DNA of DXWR (Fig. 1(a)). The success rate was more than 44.3% for pear (Yue et al., Reference Yue, Liu, Zong, Teng and Cai2014) and 53.7% for Pinus dabeshanensis (Xiang et al., Reference Xiang, Zhang, Wang, Zhang and Wu2015), but lower than 80% for pigeonpea (Dutta et al., Reference Dutta, Kumawat, Singh, Gupta, Singh, Dogra, Gaikwad, Sharma, Raje, Bandhopadhya, Datta, Singh, Bashasab, Kulwal, Wanjari, Varshney, Cook and Singh2011) and 78.2% for radish (Zhai et al., Reference Zhai, Xu, Wang, Cheng, Chen, Gong and Liu2014). The lack of amplification could be because some flanking primer pairs were designed across a splice site or chimeric cDNA sequence (Varshney et al., Reference Varshney, Grosse, Hähnel, Siefken, Prasad, Stein, Langridge, Altschmied and Graner2006). Additionally, to investigate whether these genic SSR markers developed for DXWR could be applied to commercial cultivars, we amplified fragments from the genomes of 15 cultivars using the 18 successfully amplified markers. These markers displayed a high level of polymorphism between DXWR and those cultivars (Fig. 1(b)). The results suggested that the genic SSR markers developed from DXWR could be widely applicable as molecular markers to cultivared rice.
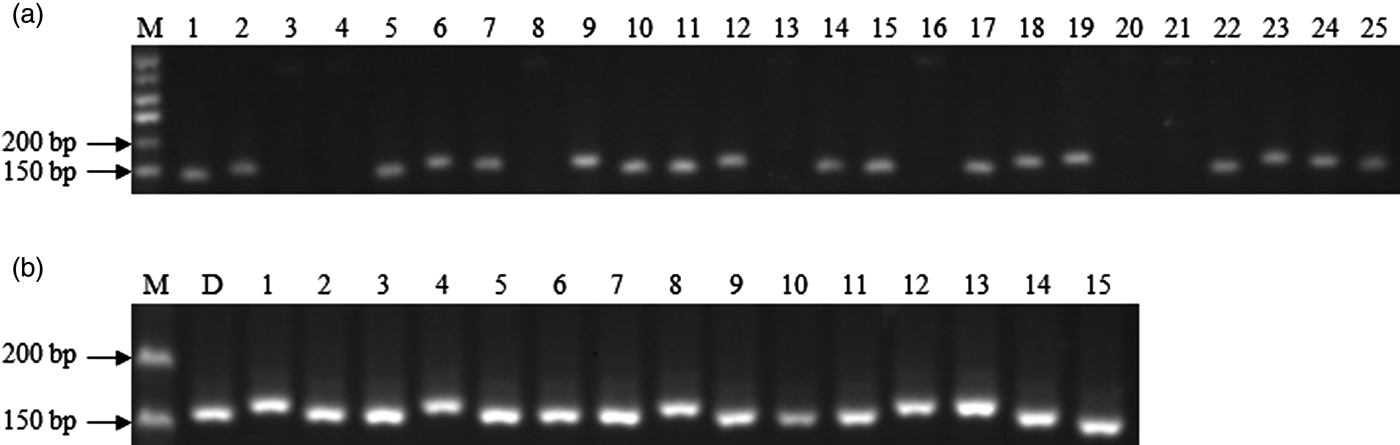
Fig. 1. (a) Electrophoresis results of 25 randomly selected primer pairs with genomic DNA of DXWR, M: DNA marker; 1–25: primer pair DgSSR29, DgSSR71, DgSSR90, DgSSR131, DgSSR206, DgSSR255, DgSSR271, DgSSR323, DgSSR628, DgSSR652, DgSSR721, DgSSR806, DgSSR879, DgSSR933, DgSSR1007, DgSSR1068, DgSSR1115, DgSSR1334, DgSSR1526, DgSSR1716, DgSSR1905, DgSSR1954, DgSSR2421, DgSSR2468, DgSSR2857. (b) Electrophoresis result of primer pair DgSSR271 in 16 rice accessions. M: DNA marker; D: Dongxiang wild rice; 1–15: 15 commercial cultivars of cultivated rice.
In summary, a comprehensive set of genic SSR markers was developed to greatly enrich the number of molecular markers available in DXWR, which will provide new opportunities to make good use of this valuable and endangered wild rice germplasm.
Supplementary Material
The supplementary material for this article can be found at https://doi.org/10.1017/S1479262116000332.
Acknowledgements
This research was partially supported by the National Natural Science Foundation of China (Grant numbers 31201191 and 31360327), the Natural Science Foundation of Jiangxi Province, China (Grant number 20142BAB204012), the Key Projects of Jiangxi Education Department (Grant number KJLD12059) and the Foundation of Jiangxi Educational Committee (Grant number GJJ14248).