Introduction
Genomic imprinting is an epigenetic regulatory phenomenon that employs both repressing and activating mechanisms to facilitate parental-specific gene expression (Bartolomei, Reference Bartolomei2009). Imprinted genes play significant roles in embryogenesis, placental formation, postnatal growth, and brain development. The developmental failure of uni-parental (bi-maternal or bi-paternal) embryos demonstrates the functional importance of imprinted genes in normal development (Ferguson-Smith & Surani, Reference Ferguson-Smith and Surani2001; Kono et al., Reference Kono, Obata, Wu, Niwa, Ono, Yamamoto, Park, Seo and Ogawa2004). Genomic imprints undergo a process of establishment, maintenance and erasure in the development of mammal. Imprints are erased in the germline, and then established during gametogenesis, but follow very different paths depending on parental origin, prenatally in pro-spermatogonia and postnatally during oocyte maturation (Barlow & Bartolomei, Reference Barlow and Bartolomei2014). Following fertilization, a global cascade of DNA demethylation is evident in the preimplantation embryos of a number of mammalian species including cattle (O’Doherty et al., Reference O’Doherty, O’Shea and Fair2012). However, in contrast with most genomic regions, imprinting control regions are protected from global demethylation processes and are maintained in the zygote and somatic cells. Proper imprinting maintenance mechanisms are highly correlated with developmental potential, but many studies have indicated that maintenance of methylation imprints is deficient during somatic cell nuclear transfer (SCNT).
Tripartite motif-containing 28 (TRIM28), also known as KRAB domain-associated protein 1 (KAP1) or transcriptional intermediary factor 1 beta (TIF1β), was discovered as an interacting protein for Krüppel-associated box zinc finger proteins (KRAB-ZFPs) (Abrink et al., Reference Abrink, Ortiz, Mark, Sanchez, Looman, Hellman, Chambon and Losson2001). TRIM28 has been reported in the regulation of many aspects of physiology, such as cell growth and differentiation (Chen et al., Reference Chen, Chen, Kurtyka, Rawal, Fulp, Haura and Cress2012), immune response (Chikuma et al., Reference Chikuma, Suita, Okazaki, Shibayama and Honjo2012), DNA damage (Lee et al., Reference Lee, Goodarzi, Adelmant, Pan, Jeggo, Marto and Chowdhury2012), tumorigenesis, embryonic development (Messerschmidt et al., Reference Messerschmidt, de Vries, Ito, Solter, Ferguson-Smith and Knowles2012), regulating Pol II activity (Bunch et al., Reference Bunch, Zheng, Burkholder, Dillon, Motola, Birrane, Ebmeier, Levine, Fargo, Hu, Taatjes and Calderwood2014), and sex-specific lethality. TRIM28 is the central scaffolding element in the heterochromatin-inducing complex containing multiple chromatin-remodeling factors. It epigenetically regulates gene expression through multiple transcriptional co-repressor complexes. TRIM28 recruits DNMT1, the H3K9me3-catalyzing histone methyltransferase SETDB1, the nucleosome remodeling and histone deacetylation (NuRD) complex, and heterochromatin protein 1 (HP1) to form transcriptional co-repressor complexes (Schultz et al., Reference Schultz, Friedman and Rauscher2001; Schultz et al., Reference Schultz, Ayyanathan, Negorev, Maul and Rauscher2002; Ivanov et al., Reference Ivanov, Peng, Yurchenko, Yap, Negorev, Schultz, Psulkowski, Fredericks, White, Maul, Sadofsky, Zhou and Rauscher2007; Wolf & Goff, Reference Wolf and Goff2007; Iyengar & Farnham, Reference Iyengar and Farnham2011;). The complex achieves DNA-binding specificity through TRIM28 interactions with KRAB-ZFPs, such as ZFP57 (Zuo et al., Reference Zuo, Sheng, Lau, McDonald, Andrade, Cullen, Bell, Iacovino, Kyba, Xu and Li2012). As a transcriptional co-repressor factor, maternal TRIM28 has been shown to protect imprinted methylation sites during preimplantation development (Messerschmidt et al., Reference Messerschmidt, de Vries, Ito, Solter, Ferguson-Smith and Knowles2012). TRIM28 derived from the oocyte is essential for the maintenance of imprinted methylation during the first cell divisions prior to the production of zygotic TRIM28, as indicated by the partial embryonic lethality resulting from maternal TRIM28 deficiency in mice (Messerschmidt et al., Reference Messerschmidt, de Vries, Ito, Solter, Ferguson-Smith and Knowles2012). However, the imprinting maintenance function of TRIM28 in the development of bovine preimplantation embryos has not been elucidated. In addition, whether the process of SCNT results in loss of maternal nuclear TRIM28, leading to subsequent failure to maintain methylation imprints in the SCNT embryos, is not known. In this study, we compared the TRIM28 expression of IVF embryos and SCNT-derived preimplantation embryos. Furthermore, we selected a specific siRNA of TRIM28 to investigate the maintenance of methylation imprints in bovine preimplantation embryos.
Materials and methods
Chemicals
All chemicals were purchased from Sigma-Aldrich Chemical Company (St Louis, MO, USA), unless otherwise stated.
siRNA transfection
Three TRIM28 (NM_001206809.1) siRNAs (si647, si742, and si1153) and a nonsense siRNA oligonucleotide (Table 1) were chemically synthesized by GenePharma Co., Ltd (Shanghai, China). In addition, the three siRNAs were mixed (siMix). Nonsense siRNA was included as a negative control. Transfection was carried out according to the instructions cited by Lipofectamine 3000 and GenePharma. The transfection efficiency of different ratios of siRNA and the Lipofectamine 3000, as well as the siRNA concentrations, were determined using a fluorescein-labelled nonsense siRNA. Cells were passaged and plated in 24-well plates before transfection at 70–80% confluence. The ratio of Lipofectamine 3000 to siRNAs was 3:1 (1.5 μl:0.5 μg) per well. Before transfection, the mixture was placed at room temperature for 15 min then added to the cells, incubated for 6 h, and the medium was changed. Fluorescence could be detected using a fluorescence microscope at 24 h after siRNA transfection. Cells were collected after 48 h for relative quantification by reverse-transcription polymerase chain reaction (RT-PCR).
Table 1 Sequence of primers and siRNA
In vitro maturation of oocytes and siRNA microinjection
Bovine ovaries were collected from a local abattoir. The procedure of in vitro maturation of oocytes was carried out according to methods established in our laboratory (Wang et al., Reference Wang, ZhaoB, Zhang, Zhang, Guan, Ma, Yin, Zhang, Tang and Li2011). The ovaries were washed three times with 0.9% (w/v) NaCl. Cumulus–oocyte complexes (COCs) were aspirated from follicles (2–8 mm) using an 18-gauge needle attached to a 10 ml syringe. For siRNA microinjection, the COCs were digested in 0.2% hyaluronidase for 3 min at 38.5°C. When all cumulus cells were separated from oocytes, 10 pl of the siRNA mixture (0.264 mg⁄ml) and nonsense RNA were microinjected into the germinal vesicle (GV)-stage oocytes using the Eppendorf CellTram vario system (Eppendorf, Hamburg, Germany). For the maturation of oocytes, COCs with intact and unexpanded cumulus cells (control), oocytes which removed cumulus microinjected simix and nonsense RNA were cultured for 22 h at 38.5°C in maturation medium in a humidified 5% CO2 incubator. The maturation medium (pH 7.2) consisted of bicarbonate-buffered TCM-199 (Gibco) supplemented with NaHCO3, 10% FBS, 1 μg/ml follicle-stimulating hormone (FSH), and 10 μg/ml luteinizing hormone (LH).
In vitro fertilization
Matured oocytes were used for in vitro fertilization (IVF). The procedure of IVF was carried out according to methods established in our laboratory (Zhang et al., Reference Zhang, Tang, Fan, Shi, Zhang, Sun and Li2016a). Frozen semen was thawed and washed three times in Dulbecco’s phosphate-buffered saline (D-PBS). It was then washed in TALP [NaCl 0.67 mg/ml, KCl 0.024 mg/ml, NaH2PO4 0.005 mg/ml, CaCl2·2H2O 0.029 mg/ml, MgCl2·6H2O 0.01 mg/ml, glucose 0.09 mg/ml, sodium pyruvate 0.01 mg/ml, 0.19 μl sodium lactate (60% v/v), NaHCO3 0.21 mg/ml, and bovine serum albumin (BSA) 0.3 mg/ml] by centrifugation. Matured oocytes (up to 20 per drop) were transferred into 80 μl TALP drops containing 106 motile sperm/ml. Coincubation was carried out for 20–22 h at 38.5°C and 5% CO2. Putative zygotes were then washed in SOF medium and were cultured in SOF medium drops. Culture medium was changed every 2 days. Embryos (from 2-cell stage to blastocysts) were collected at each developmental stage.
Bisulfite sequencing
Bisulfite sequencing was used to analyze the methylation status of our samples, as previously described (Zhang et al., Reference Zhang, Tang, Fan, Shi, Zhang, Sun and Li2015). Different stage IVF embryos (100 2-cell embryos, 50 4-cell embryos, 30 8-cell embryos, or 10 blastocysts) were treated with lysis solution (10 mM Tris–HCl, pH 7.6; 10 mM EDTA; 1% SDS; and 20 mg/ml of proteinase K) for 2 h at 37°C. The samples were then boiled for 5 min and chilled on ice. A volume of 4 μl of 2 M NaOH was added and incubated for 15 min at 50°C. Samples were mixed with two volumes of 2% low melting point agarose and pipetted into chilled mineral oil to form beads. The beads were then treated with freshly prepared bisulfite solution (2.5 M sodium metabisulfite and 125 mM hydroquinone, pH 5) for 5 h in the dark and covered with mineral oil at 50°C. The reactions were stopped by equilibration against 1 ml Tris–EDTA buffer (pH 8.0) for 4 × 15 min. After desulfonation in 0.5 ml 0.2 M NaOH for 2 × 15 min, the beads were washed with 1 ml Tris–EDTA buffer for 3 × 10 min and H2O for 2 × 15 min, and then used for PCR. The bisulfite -modified DNA PCR process were as follows: 95°C 5 min, followed by 15 cycles of 95°C 30 s and 60°C 30 s &QJ;(minus 1°C per cycle), 72°C for 1 min, then 15 cycles of 95°C 30 s and 45°C 30 s, 72°C 1 min, and a final extension step of 72°C for 7 min. PCR products were verified by electrophoresis on a 2% agarose gel, gel extraction using a Gel Extraction Kit (OMEGA), connected with the T vector (TaKaRa), pick up 15 clones for sequencing.
RNA isolation, cDNA preparation, and RT-PCR
The RNA from bovine fetal fibroblast cells, oocytes, and IVF embryos were extracted using the RNeasy Mini kit (Qiagen, Hilden, Germany). The cDNA was synthesized using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech AT311-03, Beijing, China). 18s rRNA was used as an internal control. The primers for TRIM28 and the 18s rRNA are listed in Table 1. The real-time PCR mix (25 μl) consisted of 2 μl of cDNA, 12.5 μl of SYBR green master mix, 9.5 μl of RNase-free water, and 0.5 μl each of primers (10 pmol). A reaction mix was formulated for the sample and for a no-reverse transcriptase control reaction. The program used for the amplification consisted of a denaturing cycle of 3 min at 95°C, 40 cycles of PCR (95°C for 10 s, 55°C for 45 s, and 95°C for 1 min), a melting curve analysis consisting of 95°C for 1 min followed by 55°C for 1 min, and a step cycle starting at 55°C for 10 s with a 0.5°C⁄s transition rate, and cooling at 4°C. Relative gene expression data were analyzed using real-time quantitative PCR and the 2-ΔΔCT method.
Immunofluorescence staining for H3K9me3
Immunofluorescence staining was carried out according to methods established in our laboratory (Zhang et al., Reference Zhang, Tang, Fan, Shi, Zhang, Sun and Li2016a). Zona pellucida (ZP) of bovine embryos were removed by treatment with 0.5% (w/v) Pronase in TCM-199 at 38.5°C for 5 min, washed in PBS, and fixed in 4% (w/v) paraformaldehyde for 30 min at 37°C. After fixation, the embryos were washed in PBS and permeabilized in PBS containing 0.1% (v/v) Triton X-100 for 20 min. To block non-specific binding sites, the samples were incubated for 1 h at 37°C in PBS containing 0.01% (w/v) Tween 20 and 2% (w/v) BSA, followed by an incubation in blocking solution together with primary antibody raised against H3K9me3 (dilution 1:200 v/v; Abcam) for 12 h at 4°C. The embryos were washed in blocking solution and stained with Alexa Fluor 488 goat anti-rabbit (1:200 v/v; Invitrogen) for 1 h at 37°C. DNA was stained with 10 µg/ml Hoechst 33342 for 10 min. Then, the samples were observed under a fluorescence microscope. All images were captured with the same exposure times and microscope settings. H3K9me3 levels in samples were quantified by a fluorescence intensity assay using Image-Pro Plus 6.0 software.
Fibroblast cell preparation and cryopreservation
Bovine fetal fibroblast cells were obtained from a 45-day-old fetal calf. Fetal skin tissues were cleaned with sterilized water, dissected into 1 mm pieces, and washed three times in D-PBS supplemented with 1% penicillin and streptomycin. The pieces of tissue were placed in a conical tube with Dulbecco’s modified Eagle medium (DMEM) containing 0.5% of collagenase type I in 5% CO2 at 38.5°C for 3 h. DMEM with 10% FBS (fetal bovine serum) was added to the medium to deactivate the enzyme. The cell suspension was then centrifuged at 1,000 g for 10 min. Cells were resuspended in DMEM supplemented with 10% FBS and cultured under 5% CO2 and 100% humidity at 38.5°C. Fibroblast cells were frozen and thawed as needed. To freeze the cells, the fibroblasts were resuspended in DMEM supplemented with 10% dimethyl sulfoxide (DMSO) and cooled gradually. Once they reached –80°C, they were moved to liquid nitrogen for long-term storage.
Bovine SCNT
Bovine fetal fibroblast cells (BEFs) were used as donor cells for SCNT. The SCNT protocol was performed as previously described (Zhang et al., Reference Zhang, Chen, Wang, An, Tang, Zhang, Sun and Li2016b). Briefly, oocytes with the first polar body were transferred into TCM-199-HEPES medium containing 7.5 μg/ml cytochalasin B, and then the first polar body and surrounding cytoplasm were removed using a beveled pipette. A single cell was individually transferred to the perivitelline space of the recipient cytoplasts. Cell fusion was performed using two direct current pulses of 1.2 kV/cm for 10 μs by an Electro Cell Manipulator 2001 (BTX, San Diego, CA, USA) in 0.27 M mannitol, 0.1 mM CaCl2, 0.1 mM MgCl2, and 0.05% BSA. Fused eggs were activated with 5 μM ionomycin for 5 min followed by treatment with 2 mM 6-DMAP in SOF medium (108 mM NaCl, 7.2 mM KCl, 0.3 mM KH2PO4, 5 mM NaHCO3, 3.3 mM sodium lactate, 0.07 mM kanamycin monosulfate, 0.33 mM pyruvate, 1.7 mM CaCl2·2H2O, 0.3% (w/v) fatty acid-free bovine albumin, 1% (v/v) non-essential amino acid, and 2% (v/v) essential amino acid) containing 10% FBS for 4 h at 38.5°C in 5% CO2 and 95% humidified air, then washed in SOF three times and cultured under mineral oil in droplets of SOF.
Statistical analysis
Data were analyzed with Statistics Production for Service Solution (Version 17.0; SPSS, Chicago, IL, USA) by one-way analysis of variance (ANOVA). A value of P<0.05 was considered to be significantly different.
Results
TRIM28 siRNA selection
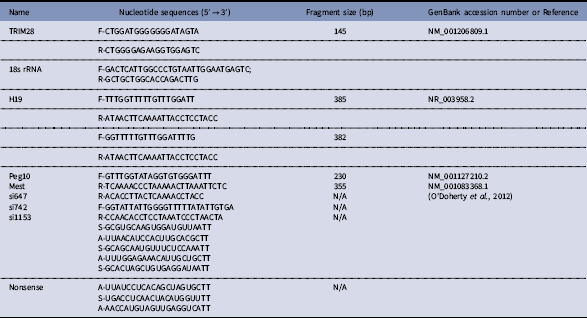
The siRNAs (si647, si742, si1153, and siMix) and nonsense control siRNA were transfected into bovine fibroblast cells by Lipofectamine 3000 and Trim28 expression was detected 48 h after transfection by RT-PCR. Transfection with siMix resulted in 82.9% knockdown of Trim28 compared with the control group and had the highest RNA interference (RNAi) efficiency among the four groups at the mRNA level. siMix and the nonsense control siRNA (control group) were microinjected into GV-stage oocytes. After culturing for 24 h, metaphase II (MII) stage oocytes were collected, and the RNAi efficiency was examined by quantitative real-time PCR. Expression of Trim28 mRNA in siMix-injected MII oocytes was significantly reduced (down-regulated 80%) compared with controls, indicating successful Trim28 knockdown by RNAi (Fig. 1A, B). Importantly, the oocyte maturation rate of the siMix microinjection group did not decrease significantly compared with the control group and nonsense RNA microinjected group (P>0.05) (Table 2).
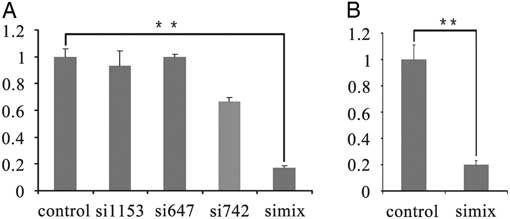
Figure 1 Real-time PCR of TRIM28 knockdown in bovine fetal fibroblast cells and oocytes. (A) Real-time PCR of TRIM28 knockdown in bovine fetal fibroblast cells at 48 h after si1153, si647, si742, and siMix were transfected. (B) Twenty-four hours after nonsense RNA (control) and siMix microinjection, Trim28 of bovine MII stage oocytes was down-regulated in the siMix microinjection group.
Table 2 Oocyte maturation rates following siMix microinjection compared with nonsense siRNA microinjected group and the un-injected group

The oocyte maturation rate of the siMix microinjection group decreased greatly, but the change was not statistically significant.
Impact of Trim28 knockdown on imprint methylation status in bovine preimplantation embryos
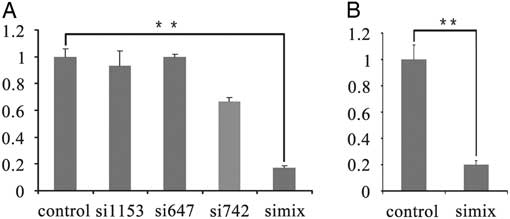
To directly investigate whether TRIM28 is involved in the maintenance of methylation imprints in bovine preimplantation embryos, the methylation status of imprinted genes was examined in the RNAi-mediated knockdown embryos. The selected TRIM28 siRNA or the negative control siRNA were injected into GV-stage oocytes. After culturing for 24 h, IVF was conducted with the TRIM28 knockdown MII oocytes, and the nonsense siRNA-injected group was used as the control. Different stage TRIM28 knockdown embryos were collected at various culturing periods, and the methylation status of the imprinted genes (H19, Mest, and Peg10) was determined by bisulfate-sequencing PCR (BSP) (Fig. 2A). TRIM28 knockdown in oocytes had a great effect on H19: H19 methylation was 1.4% (almost completely absent) in the TRIM28 knockdown embryos at the 2-cell stage, while the methylation was 74% in the IVF control at this stage. At the 2-cell stage, Trim28 mRNA was down-regulated 72.4% in the TRIM28 knockdown embryos and significantly decreased compared with the IVF control (P<0.05) (Fig. 2B), suggesting an important role for TRIM28 in the effective maintenance of methylation imprints during the development of bovine preimplantation embryos. From the 4-cell to the 8-cell stage H19 methylation increased slightly (from 23.2%to 38.7%) in the TRIM28 knockdown embryos but was significantly lower than the IVF control groups (methylation was 68.4% and 68.3% at the 4-cell and 8-cell stages, respectively). However, in the 4-cell and the 8-cell stage, the Trim28 mRNA was not significantly different between TRIM28 knockdown embryos and IVF embryos. The methylation levels of Mest and Peg10 were not significantly different in TRIM28 knockdown embryos and the control group (Fig. 4A). Importantly, the ratio of embryos that had developed to the 2-cell (cleavage rate), 4-cell, and 8-cell stages were significantly different between the TRIM28 knockdown group and the control groups (both the IVF control which matured with cumulus and the nonsense injected control) (P<0.05). For the control groups, compared with un-injected IVF control (matured with cumulus), the development rate decreased a lot in all stage in the nonsense injected control (Table 3). In this study, we did not obtain blastocysts in the TRIM28 knockdown group.
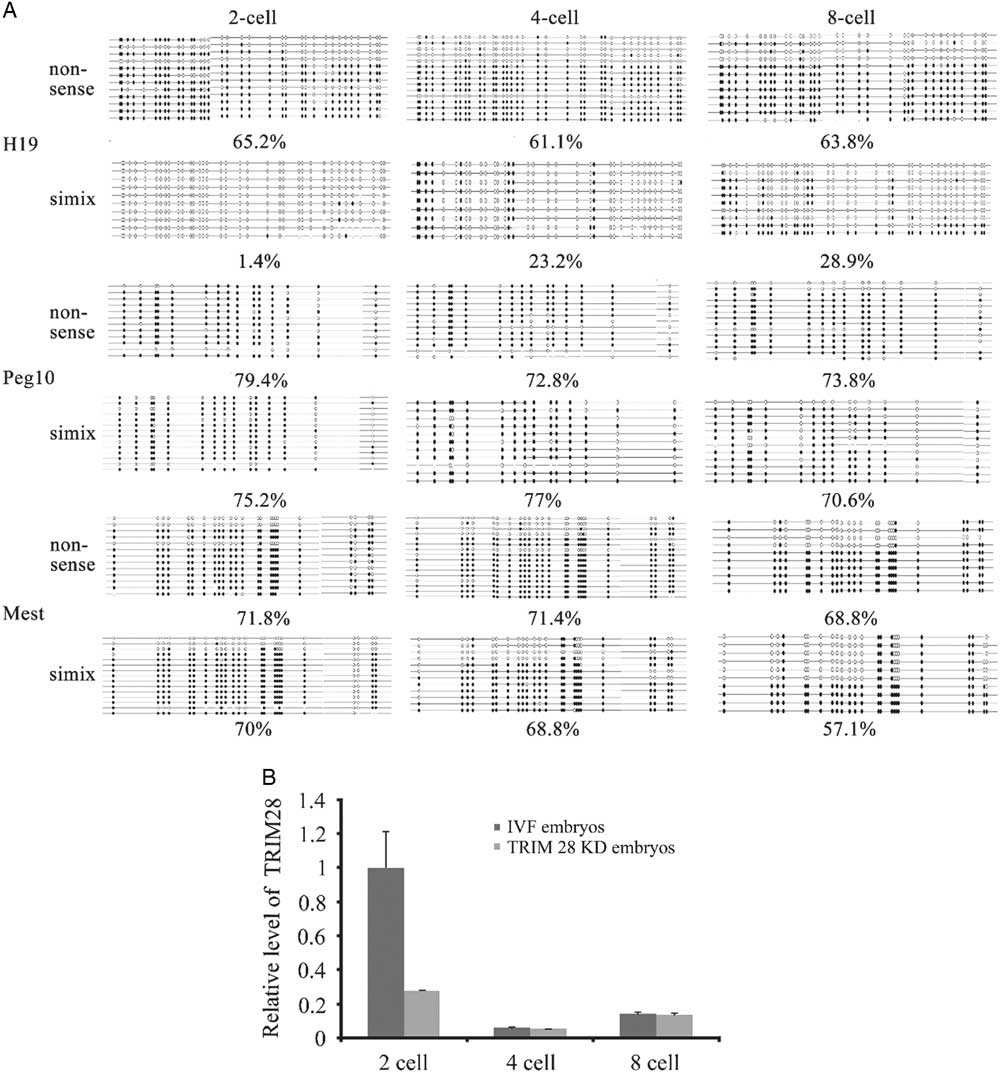
Figure 2 TRIM28 knockdown in bovine preimplantation embryos. (A) Loss of methylation at DMRs in TRIM28 knockdown bovine preimplantation embryos. TRIM28 knockdown from oocyte has the most effect on H19, which was demethylated completely at the 2-cell stage. From the 4-cell stage to the 8-cell stage the H19 methylation increased slightly but was significantly lower than the IVF control. (B) Trim28 mRNA abundance was detected in different stages of TRIM28 knockdown embryos and IVF embryos. Trim28 mRNA was significantly decreased at the 2-cell stage compared with the IVF control group (P<0.05). In the 4-cell and 8-cell stages, Trim28 mRNA was not significantly different between TRIM28 knockdown embryos and IVF embryos.
Table 3 Embryos developed to the 2-cell, 4-cell and 8-cell stages following siMix microinjection compared with the nonsense siRNA microinjected group and the no -injected group

The rates of embryos that developed to the 2-cell (cleavage rate), 4-cell, and 8-cell stages were significantly different between the siMix microinjection group and the control groups (both the IVF control which matured with cumulus and the nonsense injected control) (P<0.05). For the control groups, compared with non-injected IVF control (matured with cumulus), the development rate decreased considerably in all stages in the nonsense injected control.
Impact of TRIM28 knockdown on H3K9me3 levels in bovine preimplantation embryos
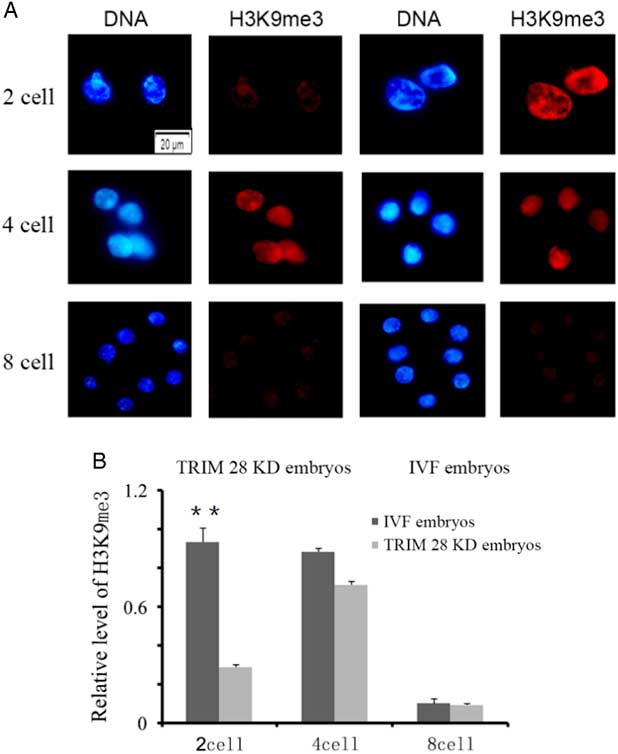
The levels of global H3 lysine 9 trimethylation (H3K9me3) were measured in TRIM28 knockdown embryos and IVF embryos at the 2-cell, 4-cell and 8-cell stages (Fig. 3A). No signals were detected in embryos stained without primary or secondary antibodies. As shown in Fig. 3A, strong H3K9me3 signals were observed at the 2-cell and 4-cell stages, but weak signals were found at the 8-cell stage during bovine IVF preimplantation embryo development. The global H3K9me3 levels at the 2-cell stage were significantly higher in the IVF group than those in the TRIM28 knockdown group (P<0.05) (Fig. 3B). No significant differences in H3K9me3 levels were observed between the IVF and TRIM28 knockdown groups at the 4-cell and 8-cell stages.
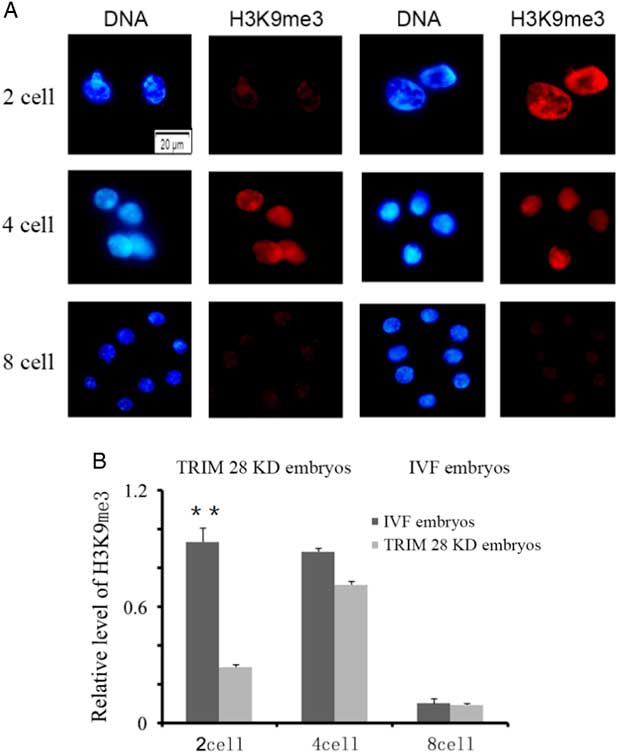
Figure 3 TRIM28 knockdown on global methylation levels of H3K9 in bovine preimplantation embryos. (A) Global methylation levels of H3K9 in embryos. Immunofluorescence staining for histone H3 lysine 9 trimethylation (red) in bovine IVF embryos and TRIM28 knockdown embryos. The DNA (blue) was stained with Hoechst 33342. (B) Quantification of H3K9me3 signal intensities in bovine IVF embryos and TRIM28 knockdown embryos.
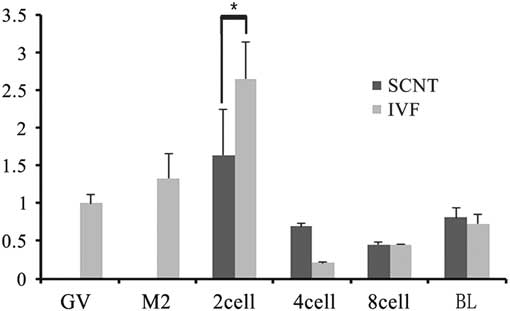
Expression of Trim28 in oocytes and preimplantation SCNT embryos
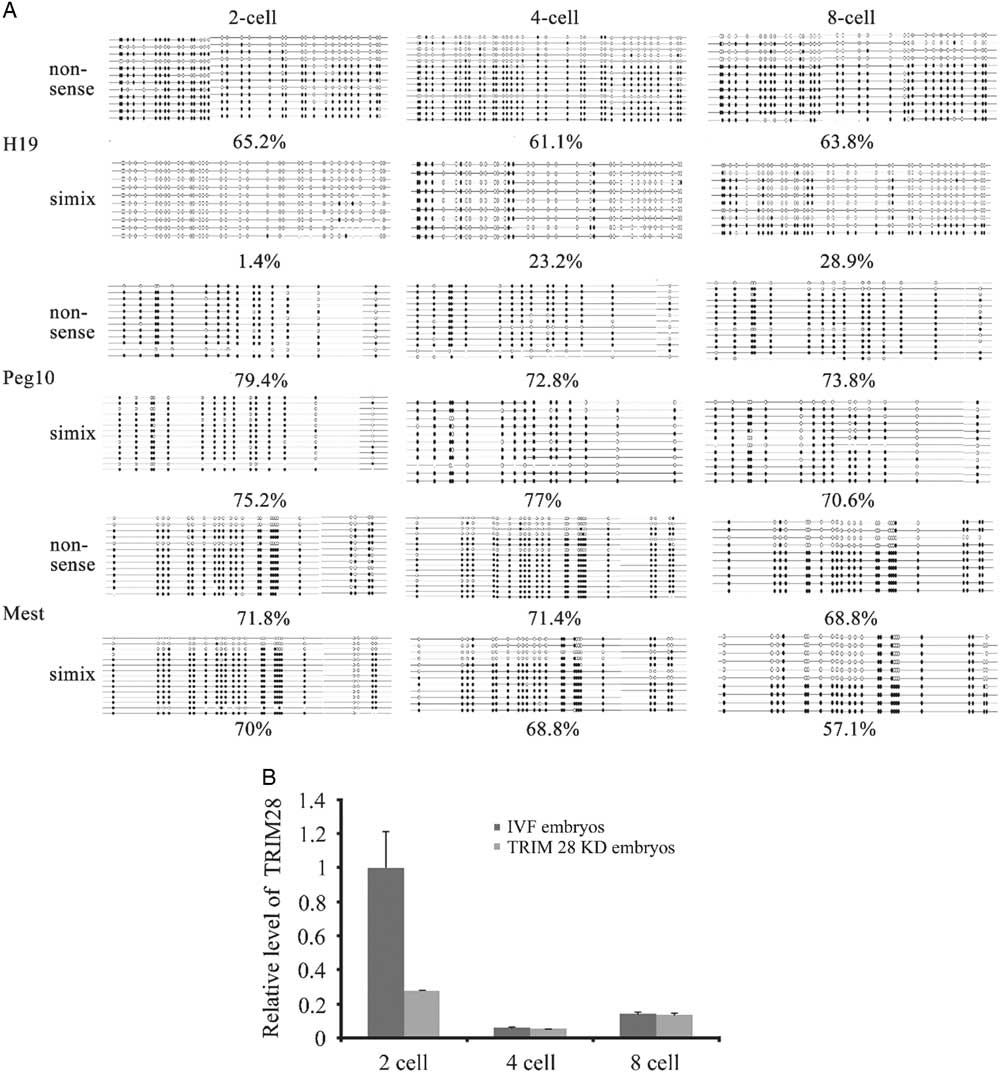
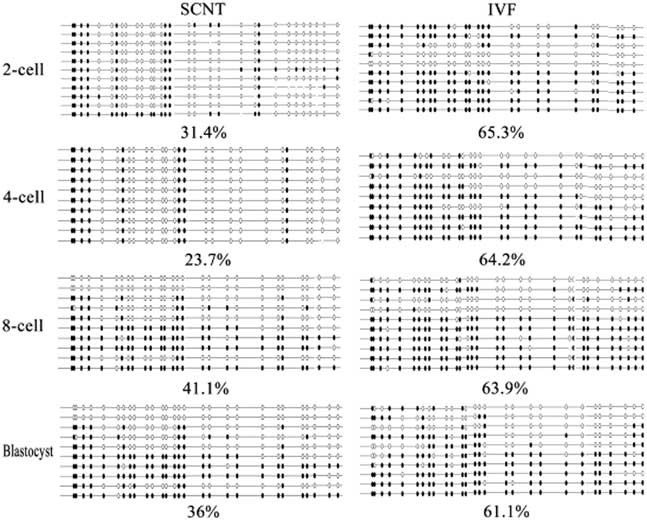
To investigate the role of TRIM28 in the maintenance of imprinted methylation during development in bovine preimplantation embryos, its mRNA abundance in oocytes (GV, MII) and different stage SCNT and IVF preimplantation embryos (2-cell, 4-cell, 8-cell, and blastocysts) were examined by RT-PCR. Quantitative RT-PCR analysis showed relatively abundant Trim28 levels in oocytes, with an increase from the GV to MII oocyte. The rise in TRIM28 during maturation supports a poly adenylation change as transcription is shut down during that period. In IVF preimplantation embryos, there was a significant increase from MII oocyte to 2-cell embryo, then a decrease at the 4-cell stage, followed by a gradual increase from the 4-cell stage to the blastocyst (Fig. 4). We then examined the expression of Trim28 in preimplantation embryos obtained from SCNT. IVF embryos had more Trim28 transcripts than SCNT embryos (P<0.05) at the 2-cell stage, but at the 4-cell stage Trim28 levels in SCNT embryos were higher than IVF embryos (P<0.05). Due to the reduction of Trim28 in SCNT embryos at the 2-cell stage, we examined the DNA methylation status of the imprinted genes H19 which is most effect by Trim28 at various developmental stages. In cloned embryos, the imprinted gene H19 was demethylated at each stage (Fig. 5), suggesting that H19 had undergone a loss of methylation during SCNT. The reduction of Trim28 may be one of the reasons which cause imprinting disorder in SCNT embryos.

Figure 4 Real-time PCR of Trim28 in oocytes and preimplantation embryos. IVF embryos had more Trim28 transcripts than SCNT embryos at the 2-cell stage (P<0.05), but at the 4-cell stage, TRIM28 levels were higher in SCNT embryos than IVF embryos (P<0.05).
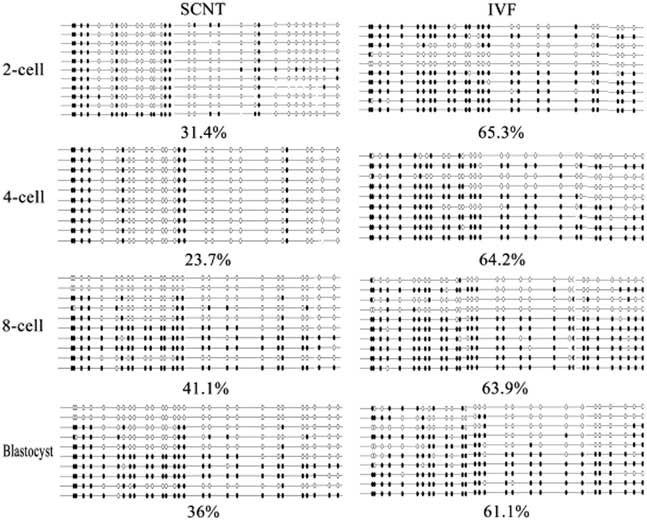
Figure 5 H19 imprint methylation status in bovine preimplantation SCNT embryos.
Discussion
TRIM28/KAP1/TIF1β is a central element in a heterochromatin-inducing macromolecular complex that was shown to protect imprinted regions from demethylation. The abundance of TRIM28 is known to be crucial for preimplantation embryo development and normal fetal development (Cammas et al., 2000; Messerschmidt et al., Reference Messerschmidt, de Vries, Ito, Solter, Ferguson-Smith and Knowles2012). In our study, we found that TRIM28 is highly expressed during oocyte maturation and persists in the preimplantation embryo, which is consistent with mouse knockout studies (Messerschmidt et al., Reference Messerschmidt, de Vries, Ito, Solter, Ferguson-Smith and Knowles2012). The highest Trim28 expression levels were seen at the 2-cell stage in both IVF and SCNT embryos. However, we observed that Trim28 expression was significantly reduced in SCNT embryos compared with IVF embryos at the 2-cell stage, suggesting that some of the maternal Trim28 is lost during SCNT. It is known that post-fertilization up to the 2-cell stage is a crucial time of DNA demethylation. TRIM28 plays an important role in protecting the genome against the processes of global DNA demethylation. In the process of SCNT, enucleation would remove a part of the transcribed fraction leaving only the novo translation-polyadenylation dependent to restore the protein in SCNT embryos. Therefore, the decrease of TRIM28 in SCNT embryos at the 2-cell stage may lead to the failure to maintain methylation imprints during the 2-cell stage in SCNT embryos.
To determine the role of TRIM28 in the imprint maintenance of bovine preimplantation embryos, IVF preimplantation embryos were collected after RNAi-mediated knockdown of Trim28, and the methylation status of the imprinted genes H19, Mest, and Peg10 were detected by BSP. The results showed that with knockdown of Trim28 in the bovine oocyte, the most affected imprinted gene was H19, which was completely demethylated at the 2-cell stage. At this stage, the Trim28 mRNA abundance of IVF embryos reached the highest level, and in the knockdown embryos Trim28 mRNA was down-regulated 72.4%. This finding indicated that after fertilization to the 2-cell stage, TRIM28 played an important role in protecting the H19 differentially methylated region (DMR) on the paternal chromosome from aberrant DNA demethylation in bovine, similar to what has been shown in mice (Messerschmidt et al., Reference Messerschmidt, de Vries, Ito, Solter, Ferguson-Smith and Knowles2012). In the TRIM28 knockdown embryos from the 4-cell to the 8-cell stage, the Trim28 mRNA reached a level similar to the IVF embryos. This may be due to the reduction of siRNA interference effects, but the regained Trim28 expression level did not rescue the loss of H19 methylation. From the 4-cell to the 8-cell stage, H19 methylation increased slightly but was significantly lower than the IVF controls, demonstrating the important role for maternal TRIM28 in protecting imprinted genes from demethylation. However, there may be other factors such as PGC7 and/or ZFP57 playing additional protective roles. Though TRIM28 binds all known imprinting control regions (Quenneville et al., Reference Quenneville, Verde, Corsinotti, Kapopoulou, Jakobsson, Offner, Baglivo, Pedone, Grimaldi, Riccio and Trono2011), the methylation levels of Mest and Peg10 were not significantly different in TRIM28 knockdown embryos and the negative control group. It is unclear why imprinted loci are not equally affected. A recent publication also showed that TRIM28 regulates paternal imprinting control regions (ICR) methylation and chromatin accessibility, with no effects on maternal ICRs (Tao et al., Reference Tao, Yen, Chitiashvili, Nakano, Kim, Hosohama, Yao, Nakano, Tan and Clark2017). But another publication showed that the amount of TRIM28 may critical for genomic imprinting because all the maternally and paternally imprinted clusters were disrupted in the zygotic Trim28 mutants (Alexander et al., Reference Alexander, Wang, Shibata, Clark and García-García2015).
TRIM28 knockdown did not affect the oocyte maturation rate. This may be because the siRNA did not affect the TRIM28 protein level for the first 24 h; however, the rate of cleavage in the 4-cell and 8-cell stages were affected significantly, which indicates that embryonic development was still controlled by the maternal protein. However, knockdown of maternal Trim28 in bovine embryos had critical effects on embryonic development. In this study, we did not obtain blastocysts, which is different from the mice and porcine knockout studies (Hamm et al., Reference Hamm, Tessanne, Murphy and Prather2014). This difference may be due to the difference in the timing of zygotic gene activation between bovine, mouse, and porcine embryos. Bovine zygotic TRIM28 is synthesized starting at the 8-cell stage and maintains the methylation imprints of cloned embryos thereafter. In addition, the poor quality of oocytes derived from the abattoir and the different cultural system may be one of the reasons for the developmental abnormalities in the TRIM28 knockdown bovine embryos. Comparing the un-injected IVF control (matured with cumulus), developmental ability of the nonsense injected group decreased greatly in all stage, suggesting that denuding GV oocytes may bring negative effects on the maturation, IVF and embryos development.
The maintenance of H3K9me3 and DNA methylation at ICRs during early embryogenesis is critical to the regulation of imprinted genes (Quenneville et al., Reference Quenneville, Verde, Corsinotti, Kapopoulou, Jakobsson, Offner, Baglivo, Pedone, Grimaldi, Riccio and Trono2011). One publication has shown that TRIM28 resists transition between somatic and pluripotent states by maintaining the H3K9me3 repressed state and keeping endogenous retroviruses (ERVs) silenced. TRIM28 knockdown resulted in dysregulation of genes nearby H3K9me3 marks (Miles et al., Reference Miles, de Vries, Gisler, Lieftink, Akhtar, Gogola, Pawlitzky, Hulsman, Tanger, Koppens, Beijersbergen and van Lohuizen2017). Here, we observed that the H3K9me3 chromatin mark decreased significantly in TRIM28 knockdown embryos at the 2-cell stage. The decrease of H3K9me3 may affect chromatin remodeling, nuclear reprogramming, and gene expression during early development, resulting in a failure of the maintenance of methylation at ICRs and a serious block in embryo development.
In conclusion, our results indicated that TRIM28 has an important role in the maintenance of the methylation status of H19 and normal bovine embryonic development, suggesting that the loss of maternal nuclear TRIM28 during SCNT may significantly contribute to the unfaithful maintenance of methylation imprints.
Financial support
This work was supported by grants from the National Natural Science Foundation in China (No. 31302047) and the Natural Science Foundation in Jilin province (20170101018JC).
Conflict of interest
No competing financial interests exist.