



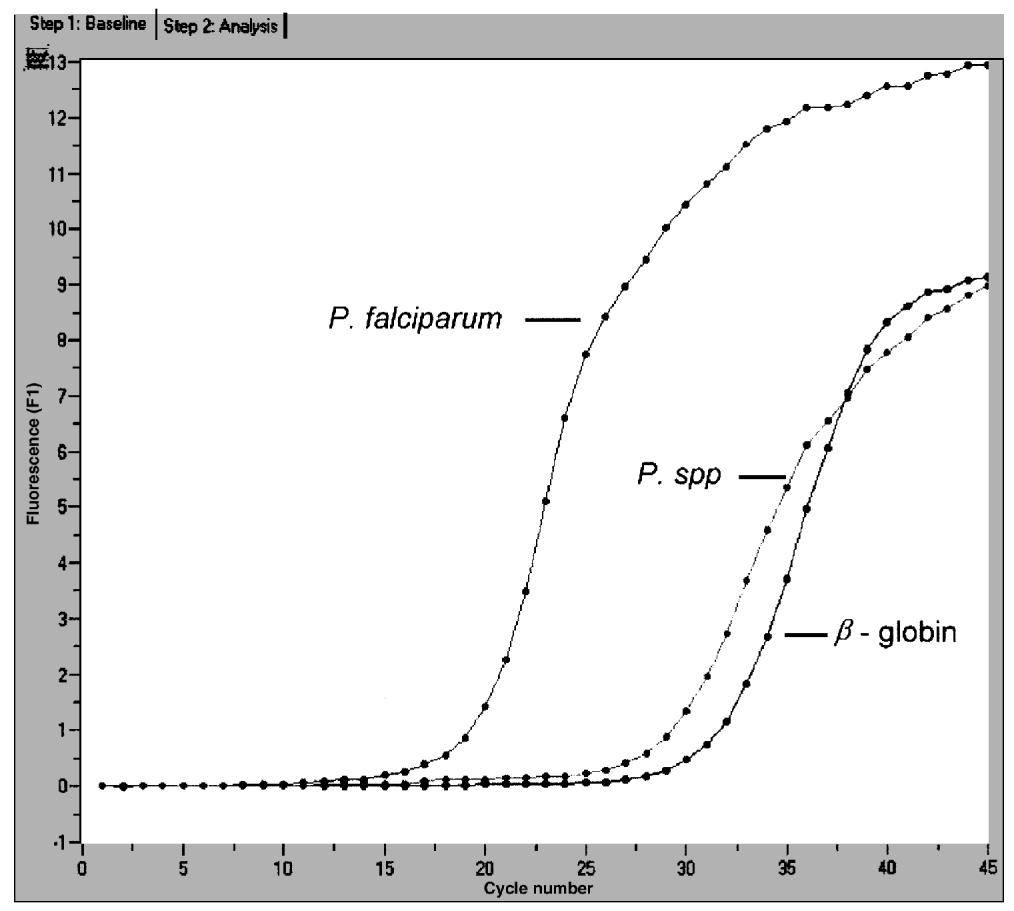


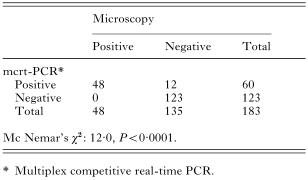



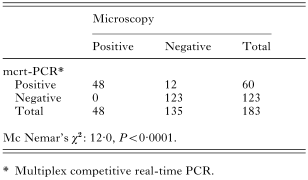

Crossref Citations
This article has been cited by the following publications. This list is generated based on data provided by Crossref.
Berry, Antoine
Vessière, Aurélia
Fabre, Richard
Benoit-Vical, Françoise
Marchou, Bruno
Massip, Patrice
and
Magnaval, Jean-François
2004.
Pfcrt K76T mutation and its associations in imported Plasmodium falciparum malaria cases.
Infection, Genetics and Evolution,
Vol. 4,
Issue. 4,
p.
361.
Bessières, M.-H.
Cassaing, S.
Berry, A.
Fabre, R.
and
Magnaval, J.-F.
2005.
Apport de la biologie moléculaire au diagnostic des parasitoses.
Médecine et Maladies Infectieuses,
Vol. 35,
Issue. ,
p.
S52.
Senescau, Alice
Berry, Antoine
Benoit-Vical, Françoise
Landt, Olfert
Fabre, Richard
Lelièvre, Joël
Cassaing, Sophie
and
Magnaval, Jean-François
2005.
Use of a Locked-Nucleic-Acid Oligomer in the Clamped-Probe Assay for Detection of a Minority
Pfcrt
K76T Mutant Population of
Plasmodium falciparum
.
Journal of Clinical Microbiology,
Vol. 43,
Issue. 7,
p.
3304.
Berry, A.
Senescau, A.
Lelièvre, J.
Benoit-Vical, F.
Fabre, R.
Marchou, B.
and
Magnaval, J.F.
2006.
Prevalence of Plasmodium falciparum cytochrome b gene mutations in isolates imported from Africa, and implications for atovaquone resistance.
Transactions of the Royal Society of Tropical Medicine and Hygiene,
Vol. 100,
Issue. 10,
p.
986.
Magnaval, J.-F.
Berry, A.
Fabre, R.
and
Cassaing, S.
2006.
Plasmodium falciparum Chloroquine-Resistance Transporter Gene Detection in Imported Plasmodium falciparum Malaria Cases.
Clinical Infectious Diseases,
Vol. 42,
Issue. 12,
p.
1806.
Erdman, Laura K.
and
Kain, Kevin C.
2008.
Molecular diagnostic and surveillance tools for global malaria control.
Travel Medicine and Infectious Disease,
Vol. 6,
Issue. 1-2,
p.
82.
Witkowski, Benoit
Iriart, Xavier
Soh, Patrice Njomnang
Menard, Sandie
Alvarez, Muriel
Naneix-Laroche, Veronique
Marchou, Bruno
Magnaval, Jean-François
Benoit-Vical, Françoise
and
Berry, Antoine
2010.
pfmdr1
Amplification Associated with Clinical Resistance to Mefloquine in West Africa: Implications for Efficacy of Artemisinin Combination Therapies
.
Journal of Clinical Microbiology,
Vol. 48,
Issue. 10,
p.
3797.
Ameri, Mehrdad
2010.
Laboratory diagnosis of malaria in nonhuman primates.
Veterinary Clinical Pathology,
Vol. 39,
Issue. 1,
p.
5.
Bourgeois, N.
Boutet, A.
Bousquet, P-J.
Basset, D.
Douard-Enault, C.
Charachon, S.
and
Lachaud, L.
2010.
Comparison of three real-time PCR methods with blood smears and rapid diagnostic test in Plasmodium sp. infection.
Clinical Microbiology and Infection,
Vol. 16,
Issue. 8,
p.
1305.
Hunt, Peter W.
2011.
Molecular diagnosis of infections and resistance in veterinary and human parasites.
Veterinary Parasitology,
Vol. 180,
Issue. 1-2,
p.
12.
Erdman, Laura K.
Hawkes, Michael
and
Kain, Kevin C.
2011.
Molecular Microbiology.
p.
691.
Polley, Spencer D.
Boadi, Samuel
Watson, Julie
Curry, Alan
and
Chiodini, Peter L.
2011.
Detection and species identification of microsporidial infections using SYBR Green real-time PCR.
Journal of Medical Microbiology
,
Vol. 60,
Issue. 4,
p.
459.
Mens, P. F.
de Bes, H. M.
Sondo, P.
Laochan, N.
Keereecharoen, L.
van Amerongen, A.
Flint, J.
Sak, J. R. S.
Proux, S.
Tinto, H.
and
Schallig, H. D. F. H.
2012.
Direct Blood PCR in Combination with Nucleic Acid Lateral Flow Immunoassay for Detection of Plasmodium Species in Settings Where Malaria Is Endemic.
Journal of Clinical Microbiology,
Vol. 50,
Issue. 11,
p.
3520.
Che, Pulin
Cui, Long
Kutsch, Olaf
Cui, Liwang
and
Li, Qianjun
2012.
Validating a Firefly Luciferase-Based High-Throughput Screening Assay for Antimalarial Drug Discovery.
ASSAY and Drug Development Technologies,
Vol. 10,
Issue. 1,
p.
61.
Boissière, Anne
Tchioffo, Majoline T.
Bachar, Dipankar
Abate, Luc
Marie, Alexandra
Nsango, Sandrine E.
Shahbazkia, Hamid R.
Awono-Ambene, Parfait H.
Levashina, Elena A.
Christen, Richard
Morlais, Isabelle
and
Vernick, Kenneth D.
2012.
Midgut Microbiota of the Malaria Mosquito Vector Anopheles gambiae and Interactions with Plasmodium falciparum Infection.
PLoS Pathogens,
Vol. 8,
Issue. 5,
p.
e1002742.
Marie, Alexandra
Boissière, Anne
Tsapi, Majoline Tchioffo
Poinsignon, Anne
Awono-Ambéné, Parfait H
Morlais, Isabelle
Remoue, Franck
and
Cornelie, Sylvie
2013.
Evaluation of a real-time quantitative PCR to measure the wild Plasmodium falciparum infectivity rate in salivary glands of Anopheles gambiae.
Malaria Journal,
Vol. 12,
Issue. 1,
Prugnolle, Franck
Rougeron, Virginie
Becquart, Pierre
Berry, Antoine
Makanga, Boris
Rahola, Nil
Arnathau, Céline
Ngoubangoye, Barthélémy
Menard, Sandie
Willaume, Eric
Ayala, Francisco J.
Fontenille, Didier
Ollomo, Benjamin
Durand, Patrick
Paupy, Christophe
and
Renaud, François
2013.
Diversity, host switching and evolution of
Plasmodium vivax
infecting African great apes
.
Proceedings of the National Academy of Sciences,
Vol. 110,
Issue. 20,
p.
8123.
Boissière, Anne
Gimonneau, Geoffrey
Tchioffo, Majoline T.
Abate, Luc
Bayibeki, Albert
Awono-Ambéné, Parfait H.
Nsango, Sandrine E.
Morlais, Isabelle
and
Snounou, Georges
2013.
Application of a qPCR Assay in the Investigation of Susceptibility to Malaria Infection of the M and S Molecular Forms of An. gambiae s.s. in Cameroon.
PLoS ONE,
Vol. 8,
Issue. 1,
p.
e54820.
Claser, Carla
Malleret, Benoit
Peng, Kaitian
Bakocevic, Nadja
Gun, Sin Yee
Russell, Bruce
Ng, Lai Guan
and
Rénia, Laurent
2014.
Rodent Plasmodium-infected red blood cells: Imaging their fates and interactions within their hosts.
Parasitology International,
Vol. 63,
Issue. 1,
p.
187.
Chua, Kek Heng
Lim, Siew Chee
Ng, Ching Ching
Lee, Ping Chin
Lim, Yvonne Ai Lian
Lau, Tze Pheng
and
Chai, Hwa Chia
2015.
Development of High Resolution Melting Analysis for the Diagnosis of Human Malaria.
Scientific Reports,
Vol. 5,
Issue. 1,