INTRODUCTION
Transposons are ubiquitous elements that comprise a large portion of the genomes of eukaryotes. Given the size of their contribution, they are considered a driving force for genome evolution and speciation (Charlesworth et al. Reference Charlesworth, Sniegowski and Stephan1994; Kidwell and Lisch, Reference Kidwell and Lisch1997). DNA transposons are a class of transposable elements that transpose without an RNA intermediate. Eukaryotic DNA transposons can be divided into 2 subclasses: Subclass 1 which contains TIR and Crypton orders and Subclass 2, which is composed of Helitron and Maverick orders (Politrons) (Feschotte and Pritham, Reference Feschotte and Pritham2007; Wicker et al. Reference Wicker, Sabot, Hua-Van, Bennetzen, Capy, Chalhoub, Flavell, Leroy, Morgante, Panaud, Paux, SanMiguel and Schulman2007). Helitrons are DNA transposons that transpose through replicative rolling-circles. Mavericks are large transposons with long Terminal Inverted Repeats (TIRs) and code for several proteins. Transposons from the TIR order use their transposase to excise double-stranded DNA for subsequent re-insertion into other regions of the genome. The TIR order, within which the present transposons were placed, can be further divided into 9 superfamilies, namely TC1; hAT; P; Mutator/Foldback; CACTA; PIF/Harbinger; Transib; piggyBac and Merlin (Wicker et al. Reference Wicker, Sabot, Hua-Van, Bennetzen, Capy, Chalhoub, Flavell, Leroy, Morgante, Panaud, Paux, SanMiguel and Schulman2007).
Schistosomes are parasitic trematodes that have several vertebrate species as definitive hosts. The genomes of Schistosoma mansoni and S. japonicum, two species with humans as their definitive host, have been recently sequenced (Berriman et al. Reference Berriman, Haas, LoVerde, Wilson, Dillon, Cerqueira, Mashiyama, Al-Lazikani, Andrade, Ashton, Aslett, Bartholomeu, Blandin, Caffrey, Coghlan, Coulson, Day, Delcher, DeMarco, Djikeng, Eyre, Gamble, Ghedin, Gu, Hertz-Fowler, Hirai, Hirai, Houston, Ivens, Johnston, Lacerda, Macedo, McVeigh, Ning, Oliveira, Overington, Parkhill, Pertea, Pierce, Protasio, Quail, Rajandream, Rogers, Sajid, Salzberg, Stanke, Tivey, White, Williams, Wortman, Wu, Zamanian, Zerlotini, Fraser-Liggett, Barrell and El-Sayed2009; Zhou et al. Reference Zhou, Zheng, Chen, Zhang, Wang, Guo, Huang, Zhang, Huang, Jin, Dou, Hasegawa, Wang, Zhang, Zhou, Tao, Cao, Li, Vinar, Brejova, Brown, Li, Miller, Blair, Zhong, Chen, Hu, Wang, Zhang, Song, Chen, Xu, Xu, Ju, Huang, Brindley, McManus, Feng, Han, Lu, Ren, Wang, Gu, Kang, Chen, Chen, Chen, Wang, Yan, Wang, Lv, Jin, Wang, Pu, Zhang, Zhang, Hu, Zhu, Wang, Yu, Wang, Yang, Ning, Beriman, Wei, Ruan, Zhao, Wang, Liu, Wang, Zheng, Zhang, Wang and Han2009). Their analysis revealed that retrotransposable elements constitute a considerable fraction, divided into several families (Venancio et al. Reference Venancio, Wilson, Verjovski-Almeida and DeMarco2010). In contrast, DNA transposons constitute a modest fraction of the genome and with only 2 previously described, one of them belonging to the Merlin superfamily (Feschotte, Reference Feschotte2004), the other being the first transposon from the CACTA superfamily described in animals (DeMarco et al. Reference DeMarco, Venancio and Verjovski-Almeida2006).
The mutator transposon was first described in Zea mays, where it is highly active, resulting in increased mutation rates in lineages that possess the element. The maize autonomous mutator element (MuDR) is 4·9 kbases long, displays 220 bp TIRs and creates 9 bp TSDs (Lisch, Reference Lisch2002). MuDR produces 2 transcripts named mudrA and mudrB which are initiated from the opposite TIRs of the elements converging to its centre, and both transcripts are spliced (Hershberger et al. Reference Hershberger, Benito, Hardeman, Warren, Chandler and Walbot1995). Mutator-like Elements (MULEs) have also been described in other plants (Lisch, Reference Lisch2002) as well as fungi (Chalvet et al. Reference Chalvet, Grimaldi, Kaper, Langin and Daboussi2003) and protists (Pritham et al. Reference Pritham, Feschotte and Wessler2005; Lopes et al. Reference Lopes, Silva, Benchimol, Costa, Pereira and Carareto2009). Recently, a new family of MULEs named Phantom was described in several metazoan organisms (Marquez and Pritham, Reference Marquez and Pritham2010) constituting the first systematic description in metazoans. All these MULEs share a conserved transposase domain containing DDE or DX34E residues followed by a CX2H motif, which is distantly related to the insertion sequence IS256 of prokaryotes (Hua-Van and Capy, Reference Hua-Van and Capy2008).
Here, we describe 2 novel families of transposons from the genomes of the human parasites S. mansoni and S. japonicum belonging to the Mutator superfamily, with distinct characteristics from Phantom, the other recently described metazoan transposon family (Marquez and Pritham, Reference Marquez and Pritham2010); these differences are the absence of TIRs, presence of 9 bp Target Duplication Sites (TSD) instead of 10 bp TSD and presence of a SWIM zinc-finger domain. We named these novel families of transposons Curupira, after the figure from South American Tupi Indian mythology, with feet that are turned backwards to create tracks that trick anyone trying to follow (or flee from) him (Smith, Reference Smith1996; Almeida and Portella, Reference Almeida and Portella2006).
MATERIALS AND METHODS
In silico detection of Curupira transposons
A search for S. mansoni transposable elements was performed using several MULE transposase amino acid sequences as queries for the S. mansoni genome (Berriman et al. Reference Berriman, Haas, LoVerde, Wilson, Dillon, Cerqueira, Mashiyama, Al-Lazikani, Andrade, Ashton, Aslett, Bartholomeu, Blandin, Caffrey, Coghlan, Coulson, Day, Delcher, DeMarco, Djikeng, Eyre, Gamble, Ghedin, Gu, Hertz-Fowler, Hirai, Hirai, Houston, Ivens, Johnston, Lacerda, Macedo, McVeigh, Ning, Oliveira, Overington, Parkhill, Pertea, Pierce, Protasio, Quail, Rajandream, Rogers, Sajid, Salzberg, Stanke, Tivey, White, Williams, Wortman, Wu, Zamanian, Zerlotini, Fraser-Liggett, Barrell and El-Sayed2009) with a stand-alone version of the tBLASTn program. After discovery of the S. mansoni Curupira-1 sequence, the amino acid sequence of its transposase domain was used for further searches in both S. mansoni and S. japonicum (Zhou et al. Reference Zhou, Zheng, Chen, Zhang, Wang, Guo, Huang, Zhang, Huang, Jin, Dou, Hasegawa, Wang, Zhang, Zhou, Tao, Cao, Li, Vinar, Brejova, Brown, Li, Miller, Blair, Zhong, Chen, Hu, Wang, Zhang, Song, Chen, Xu, Xu, Ju, Huang, Brindley, McManus, Feng, Han, Lu, Ren, Wang, Gu, Kang, Chen, Chen, Chen, Wang, Yan, Wang, Lv, Jin, Wang, Pu, Zhang, Zhang, Hu, Zhu, Wang, Yu, Wang, Yang, Ning, Beriman, Wei, Ruan, Zhao, Wang, Liu, Wang, Zheng, Zhang, Wang and Han2009) genomes again using tBLASTn.
For each transposon, the nucleotide sequence with similarity to other transposases was selected and used for BLASTn searches for additional copies in the genome. The 10 regions of the genome with the best score in the alignment had their sequences plus an additional 3000 bp of genomic sequence at each of their 2 flanking regions retrieved. The recovered sequences were aligned with the MUSCLE program (Edgar, Reference Edgar2004), and manual inspection of portions showing conservation allowed us to determine the transposons’ extremities. In cases where the 10 best alignments did not provide support for deduction of extremities, the next 5 additional sequences with highest scores were added to the alignment and the analysis was repeated. Sequences for canonical copies of SmCurupira-1, SmCurupira-2 and SjCurupira-2 were deposited as TPAs in the European Nucleotide Archive under Accession numbers BN001525-BN001527.
Transposase domain alignment and construction of phylogenetic trees
Using the deduced protein sequence of the transposase domain from the Curupira transposon, we retrieved several other sequences with significant similarity (e-values less than 10−4) in a tBLASTn search against the genomes of metazoans at NCBI. We used the sequences along with those of known MULE transposons to perform an alignment using the ClustalX2 program, followed by manual refinement. Phylogenetic analyses were performed with the maximum likelihood algorithm using the phyML 3.0 program (Guindon and Gascuel, Reference Guindon and Gascuel2003), with default parameters and LG matrix (Le and Gascuel, Reference Le and Gascuel2008). Trees were visualized and exported using the MEGA 4 package (Tamura et al. Reference Tamura, Dudley, Nei and Kumar2007).
Estimation of genomic content for DNA transposons
We estimated the genomic content of the different DNA transposon families using the same methodology employed by Venancio and coworkers (Venancio et al. Reference Venancio, Wilson, Verjovski-Almeida and DeMarco2010), which is based on element identification followed by counting the relative abundance of all shotgun reads generated by the S. mansoni and S. japonicum genome projects that map to the corresponding element.
Phylogenetic analysis of Curupira copies
The nucleotide sequences corresponding to the transposase domain (coverage >80% of the domain) for the different schistosome Curupira families were extracted from the genomes of S. mansoni and S. japonicum and a multiple alignment was performed using MUSCLE (Edgar, Reference Edgar2004), for each of the families. After manual inspection to remove sequences with large insertions/deletions, we calculated the pairwise distances between the members of each family. Phylogenetic analyses were performed with the approximately maximum likelihood algorithm using the FastTree 2.1.3 program (Price et al. Reference Price, Dehal and Arkin2010). Trees were visualized and exported using the MEGA 4 (Tamura et al. Reference Tamura, Dudley, Nei and Kumar2007) package.
RT-PCR and RACE reactions
S. mansoni adult parasites were obtained by portal perfusion of hamsters 7 to 8 weeks after infection. Worms were collected in RNALater (Ambion) according to the manufacturer's instructions. Then mRNAs were extracted using μMAC isolation kits (Miltenyi Biotec) from 40 mg of tissue and treated with RQ1 RNase-free DNase (1 U/10 μl; Promega) for 30 min at 37°C. cDNAs were prepared from 200 ng of mRNA using Superscript III (Invitrogen), following the manufacturer's instructions. PCR reactions were performed with Taq polymerase (Fermentas) and specific primers for SmCurupira-1. RACE was performed with the 5′ and 3′ RACE System for Rapid Amplification of cDNA Ends kits (Invitrogen), using specific primers for the SmCurupira-1 transposon, following the manufacturer's instructions. Representative sequences of truncated transcripts of SmCurupira-1 were deposited in the European Nucleotide Archive under Accession numbers FR693365 and FR693648. These transcript sequences and sequences from the public databases were mapped to the genomic sequences of transposons using Spidey program (Wheelan et al. Reference Wheelan, Church and Ostell2001).
RESULTS
The genomes of S. mansoni and S. japonicum display transposons of the Mutator superfamily
As a result of a transposable elements search in the S. mansoni genome sequence, we obtained a significant alignment (e-value 10−6) between a Jittery transposase sequence from a Zea mays MULE (Xu et al. Reference Xu, Yan, Maurais, Fu, O'Brien, Mottinger and Dooner2004) and a portion (bases 256352 to 256717) of the S. mansoni genomic scaffold Smp_scaff000052 (Accession number FN357343.1). Using this portion of the S. mansoni genome as query against the rest of the genome permitted us to retrieve several similar loci, indicating that it was in fact a repetitive element in the genome; we termed it SmCurupira-1. Further BLASTn alignments of regions flanking the transposase domain against the genome, and multiple alignments of the retrieved sequences, permitted us to establish the full-length sequence of the element and pinpoint 8 copies with apparently intact ends (Fig. 1). It is noteworthy that we could not detect TIRs at SmCurupira-1 extremities, as commonly seen in other MULE elements. However, the presence of TSDs of 9 base pairs flanking 6 of the 8 inspected copies (Fig. 1) appears to confirm that the extremities inferred by us are authentic.

Fig. 1. Sequence of the 5′ and 3′ ends of full-length copies of Curupira transposons. Nucleotides shaded in black indicate sequences from the ends of the Curupira transposons, nucleotides without shading in the same columns contain mutations relative to the majority of sequences. Boxed nucleotides indicate Target Site Duplications (TSD) resulting from insertion of these elements into the genomic DNA (represented without shading). Numbers on the left indicate the scaffold of the Schistosoma mansoni or S. japonicum genomes from which the sequence was obtained.
Although all of the 8 copies span most of the transposon length, some of them had internal deletions or insertions relative to the other copies or some undetermined bases in the genomic sequence (represented by Ns). Therefore, we selected for further analysis a 4878 bp long copy derived from supercontig Smp_scaff000189 (Accession number FN357480.1; bases 384776 to 389653), which was considered to be the best representative of an intact element due to the lack of insertions/deletions relative to the majority of aligned sequences; it is hereafter referred to as the canonical copy.
It was possible to identify a nucleotide sequence coding for the MULE transposase domain (pfam10551) between bases 1101 to 1394 of the SmCurupira-1 transposon. Use of this amino acid sequence as query to perform further tBLASTn searches in the S. mansoni genome resulted in the detection of a related family of elements that was termed SmCurupira-2 (Fig. 1).
Searches performed in the S. japonicum genome using SmCurupira-1 and SmCurupira-2 detected 2 related families in this organism, which were termed SjCurupira-1 and SjCurupira-2 and the extremities were identified by analyses similar to those performed for SmCurupira-1. We also confirmed that all Curupira families lack TIRs at their extremities and generate 9 bp TSD when inserted into a genomic locus (Fig. 1). It is noteworthy that the S. mansoni elements display a larger proportion of sampled full-length elements with conserved TSD (10/13) when compared to the S. japonicum elements (2/11), suggesting the prominence of recent insertions in the S. mansoni genome. Alternatively, the discrepancy in TSD abundance may reflect the fact that the S. japonicum elements may insert more frequently than the S. mansoni ones at blunt-end genomic break-points, an event which would result in transposable elements with no TSD.
Following the same rationale used for SmCurupira-1, we also chose copies to represent the canonical SjCurupira-1 (Accession number FN331292.1, bases 122640 to 127162) and SjCurupira-2 (Accession number FN331748.1, bases 22466 to 27672) elements. Curiously, all copies of SmCurupira-2 with intact extremities displayed in Fig. 1 did not have a transposase domain in their sequence, whereas all 5 copies of SmCurupira-2 with an integral transposase domain displayed at least 1 degraded extremity. Therefore we decided to make an in silico reconstruction of an intact autonomous SmCurupira-2 element by fusing 3866 bp from the 5′-end of a 3′-truncated transposase containing element (Accession number FN357659.1, bases 2500–6365) with 849 bp of the 3′ extremity of a non-autonomous SmCurupira-2 element (Accession number FN357382.1, bases 74052–74900). In silico reconstruction of a complete copy of this transposase provides a reference sequence for our studies and may represent a first step towards future production of an active element from fragments of inactive ones, as previously achieved by Ivics and collaborators with the Sleeping Beauty transposons (Ivics et al. Reference Ivics, Hackett, Plasterk and Izsvak1997).
Blastx searches with sequences from each of the deduced transposon families against the GenBank NR database indicate that they have limited similarity to sequences from other organisms, with best alignments to transposon proteins with e-values ranging from 10−4 to 10−14and identities from 24 to 38% (Table 1). This finding suggested that these newly detected transposons should represent a relatively divergent group in relation to the previously described MULE transposons.
Table 1. Alignment of Curupira transposons to other transposons in the GenBank NR database using Blastx program

a Best hit considering only organisms outside the Schistosoma genus.
Using the methodology previously described by us (Venancio et al. Reference Venancio, Wilson, Verjovski-Almeida and DeMarco2010), we estimated the abundance of Curupira transposons, as well as of the other previously described Merlin (Feschotte, Reference Feschotte2004), and TRC-1 transposons (DeMarco et al. Reference DeMarco, Venancio and Verjovski-Almeida2006) in the genomes of S. mansoni and S. japonicum (Table 2). The 2 Curupira transposons appeared to be the most highly represented DNA transposons in S. mansoni so far, with slightly higher representation than Merlin transposons; the same relative value was observed for the 2 families in S. japonicum. In contrast, DNA transposons of the TRC family were the less abundant with a representation corresponding to one fifth to one tenth of the other 2 families. It is noteworthy that DNA transposons have a far lower representation in both S. mansoni and S. japonicum genomes than retrotransposons, which make up a considerable fraction of both (Venancio et al. Reference Venancio, Wilson, Verjovski-Almeida and DeMarco2010).
Table 2. Representation of DNA transposons in the genomes of schistosomes (% of genome)

* Numbers in parentheses indicate abundances of Curupira-1 and -2, respectively.
Curupira transposons produce spliced transcripts
We were unable to identify a large ORF that could encode a complete transposase in any of the Curupira transposons using the ORFinder tool from NCBI. Indeed, the transposase and zinc-finger domains characteristic of mutator elements were detected in different ORFs. This led us to believe that transcripts from these elements were spliced and only a mature transcript would code for a complete transposase.
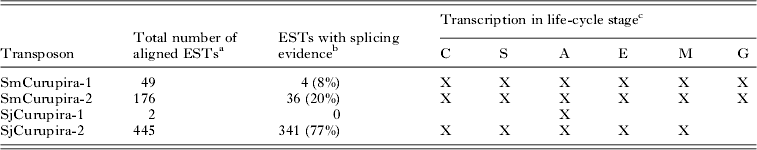
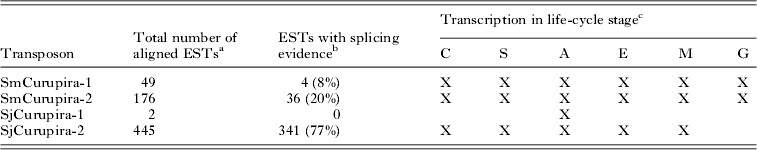
Searches with canonical Curupira elements as queries against the respective S. mansoni and S. japonicum ESTs database at NCBI resulted in several significant alignments with publicly available ESTs (e-value lower than 10−10) for most of elements except SjCurupira-1 (Table 3), providing evidence that these elements are transcribed. When ESTs were mapped to the canonical element using the Spidey program several of them displayed evidence of splicing (Fig. 2). In addition, we found several full-length sequences for SjCurupira-2 transcripts in the NR database at NCBI (Fig. 2). This indicates that these transcripts were produced from elements with intact splicing sites, which suggest they were relatively intact elements.

Fig. 2. Spliced transcripts detected in public databases. The figure shows representative examples of ESTs or RNA transcripts from public databases mapping to the following transposons: (A) SmCurupira-1, (B) SjCurupira-2 and (C) SmCurupira-2. White bars illustrate the representative transposon copies. Portions highlighted in grey indicate the region coding for the transposase domain. The hatched portion in SjCurupira-2 is similar to the previously described retrotransposon Perere-3 from S. mansoni. Wide horizontal black lines represent the exons and narrow lines the introns. Dotted narrow lines represent introns without the canonical splicing sites.
Table 3. Number of significant alignments (e-value <10−10) of Curupira transposons to schistosome transcript sequences from the NCBI EST database
a Excluding alignment with internal sequences similar to retrotransposons present in SjCurupira-2 and SmCurupira-2.
b ESTs were considered as displaying splice evidence when more than 1 exon was predicted and conserved canonical donor and acceptor splice sites were detected in at least 1 intron when mapping the EST to the canonical transposon copy using the Spidey program. Numbers in parentheses indicates percentage of total aligned ESTs.
c X indicates evidence for transcription in a given life-cycle stage determined by the presence of ESTs for that stage in the public database. C-cercaria; S-schistosomulum; A-adult worm; E-egg; M-miracidium; G-germ ball.
SjCurupira-2 displayed the largest proportion of spliced ESTs (77%), while SmCurupira-1 displayed only a small proportion of spliced ESTs (8%). It is curious that despite the large number of SmCurupira-2 ESTs found, only 1 contained the transposase domain, while the majority had a discontinuous alignment with the canonical element, in which the portion containing the transposase domain was skipped (Fig. 2). This does not appear to be the result of splicing since no canonical splicing sites were detected in the putative intron; instead it appears to be the product of transcription from truncated elements. In fact, a BLASTn search of these ESTs against the S. mansoni genome retrieved truncated copies in which the tranposase domain was absent and alignment of the cited portion of the EST was no longer discontinuous. Other portions of the ESTs from SmCurupira-2 also had a discontinuous alignment with the canonical element, and in several instances it does appear to be a product of splicing, since canonical splicing sites are detected at the extremities of the formed exons. SjCurupira-1 had only a couple of ESTs detected in adults and neither of them presented evidence of splicing (Table 3), suggesting that this transposon has a low transcription level.
We performed RT-PCR experiments to amplify transcripts for the SmCurupira-1 transposon in order to verify the apparent scarcity of spliced transcripts for this element. Amplifications using specific primers designed from regions of SmCurupira-1 that showed evidence of transcription and RACE reactions resulted in sequencing of transcripts with no evidence of splicing; several of them were derived from truncated copies (data not shown).
Searching for ESTs from SmCurupira-1 and -2 in the public database revealed the presence of transposon transcription in all of S. mansoni life-cycle stages investigated, namely cercaria, schistosomulum, adult worm, egg, miracidium, and germ ball (Table 3). ESTs for SjCurupira-2 were also detected in all S. japonicum stages except for germ balls (no ESTs are available for this life stage in the S. japonicum EST database). Overall, data from Table 3 suggest that Curupira transposons are ubiquitously transcribed throughout the life cycle of the parasite.
Curupira transposons represent a novel lineage of metazoan Mutator transposons
Curupira amino acid sequences for the transposase domain were aligned with other Mutator transposases (Fig. 3). It can be seen that all the Curupira transposons display the DX18DX15E integrase signature described for some MULE elements, in which the first and last residues correspond to the DX34E amino acid signature found in all MULE elements (Rossi et al. Reference Rossi, Araujo, de Jesus, Varani and Van Sluys2004; Hua-Van and Capy, Reference Hua-Van and Capy2008). In addition, a DX20-21CX2H motif, also characteristic of MULE transposases (Lopes et al. Reference Lopes, Silva, Benchimol, Costa, Pereira and Carareto2009), is present in Curupira transposons.

Fig. 3. Multiple alignment of domains from Curupira transposons. ClustalX2 alignments for conserved regions of transposase, WRKY/FAR1 and SWIM zinc-finger domains are shown. Shading indicates level of conservation of amino acids. Arrowheads indicate conserved residues from the DX18DX15E and DX20-21CX2H motifis characteristic of MULEs transposases.
Using the transposase domain of the SmCurupira-2 transposon as query for tBLASTn searches in the available genome and transcript databases at NCBI, we have been able to obtain alignments with identities ranging from 31 to 37% to transcripts from the platyhelminth Opisthorchis viverrini and the cnidarian Hydra magnipapillata and to genomic sequences from the mollusk Aplysia californica, and the oomycete Phytophthora infestans. These sequences were previously not annotated as transposons, thus representing novel elements.
Phylogenetic analysis of the Curupira transposase domain alignment (Fig. 4) has grouped the two Schistosoma Curupira elements along with transposase domains from O. viverrini, A. californica, H. magnipapillata and P. infestans in a monophyletic group with relatively high bootstrap support. This leads us to propose that Curupira transposons constitute a new group of MULE transposons that contains several metazoan members.

Fig. 4. Phylogenetic tree of the Mutator superfamily. A phylogenetic tree based on the transposase domain alignment of Mutator elements was constructed using maximum likelihood. Elements are represented by species names followed by the sequence GI number at GenBank. Division of the elements into families is indicated on the right of the figure. Numbers near nodes represent likelihood support values; only likelihood support values equal to or higher than 50 are shown. IS256 elements were used as the out-group.
Our analysis also indicates that the Curupira group is only distantly related (Fig. 4) to another group of transposons recently described in Metazoa, namely Phantom (Marquez & Pritham, Reference Marquez and Pritham2010), and suggests that two independent lineages of Mutator transposons were established early in the evolution of the metazoan genomes.
In addition to the transposase domain itself, other domains usually found in MULE transposons have also been detected in Curupira. All Schistosoma Curupira transposons contain a SWIM zinc-finger domain (CDD: cl11618), which has a CXCX9CXH motif and is located past the C-terminus of the transposase. We have been able to detect a WRKY/FAR1-domain in SmCurupira-1 (Fig. 3) near the protein N-terminus, which is encoded by 2 transposon exons (as evidenced by EST Accession number CD094552.1). SjCurupira-1 also displays a similar region coding for this domain. It is notable that SmCurupira-2 and SjCurupira-2 do not display a WRKY/FAR1-domain, suggesting that this domain has been lost/gained after the split between Curupira-1 and Curupira-2.
Analysis of the protein encoded by the H. magnipapillata transcript showed that a WRKY/FAR1-domain is present near the N-terminus, whereas the C-terminus of the protein encoded by this transcript appears to be truncated and no SWIM zinc-finger domain was detected. The protein encoded by the O. viverrini EST sequence appeared to be truncated and no domain additional to the transposase was detected. The sequence of proteins encoded by both A. californica and P. infestans elements displayed a SWIM zinc-finger domain. In addition, we analysed the regions 5 kbp upstream and downstream of the stretch coding the transposase domain of A. californica and P. infestans and no TIRs were detected. This suggests that most of these newly detected elements have characteristics in common with the Schistosoma Curupira transposons, providing further support for their grouping.
Curupira transposon families underwent recent expansion in both the S. mansoni and S. japonicum genomes
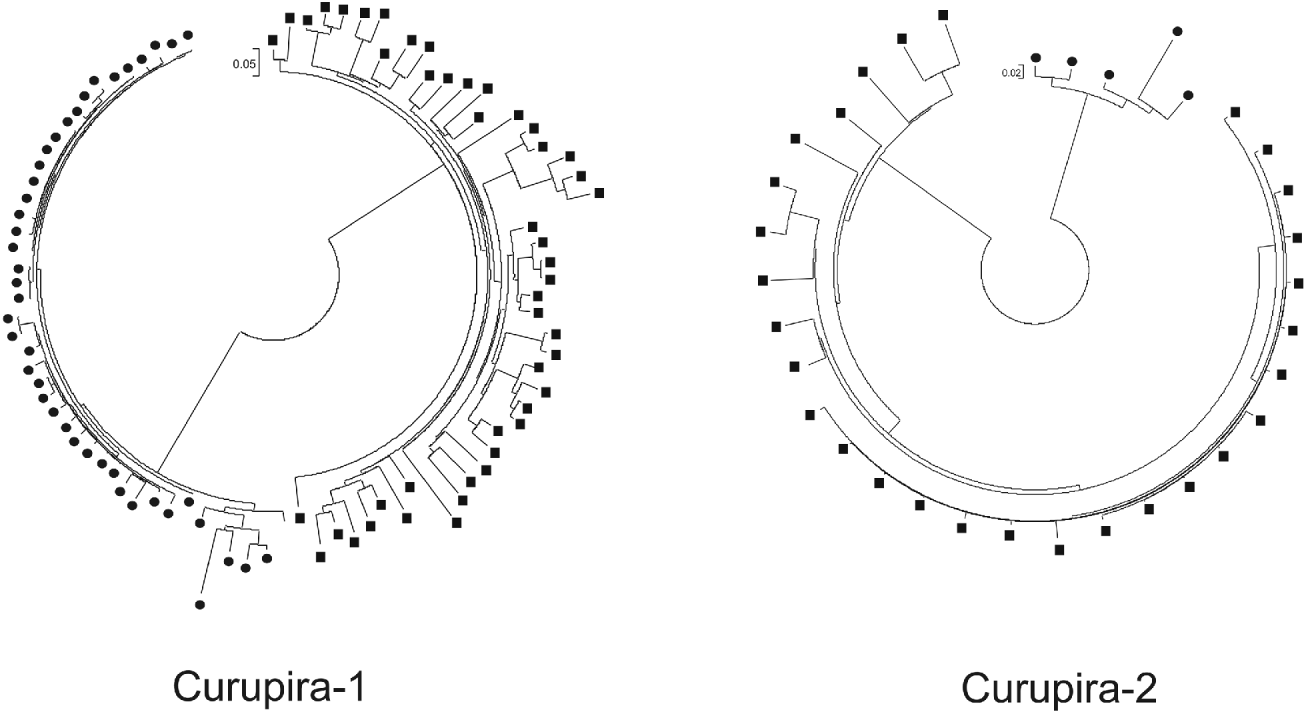
In order to study the evolutionary history of Curupira-1 and -2 in S. mansoni and S. japonicum genomes, we performed a separate multiple alignment for each of these families with different copies of nucleotide sequences encoding the transposase domain, followed by a phylogenetic analysis using the maximum likelihood algorithm. Only sequence copies that encompass at least 80% of the domain were included in this analysis. Results shown in Fig. 5 indicate that there is a very small distance between most of Curupira-1 elements in S. mansoni, suggesting that a recent expansion of this family has occurred. In contrast, copies of Curupira-1 from the S. japonicum genome are more distant to each other indicating a more ancient divergence between the members. The opposite occurs for the Curupira-2 family, in which a considerable portion of the S. japonicum elements are evolutionarily very close while the few intact copies of transposase domains from S. mansoni Curupira-2 are more distant. In fact, the Curupira-1 family is more abundant than Curupira-2 in S. mansoni (Table 2) and the opposite occurs in S. japonicum.
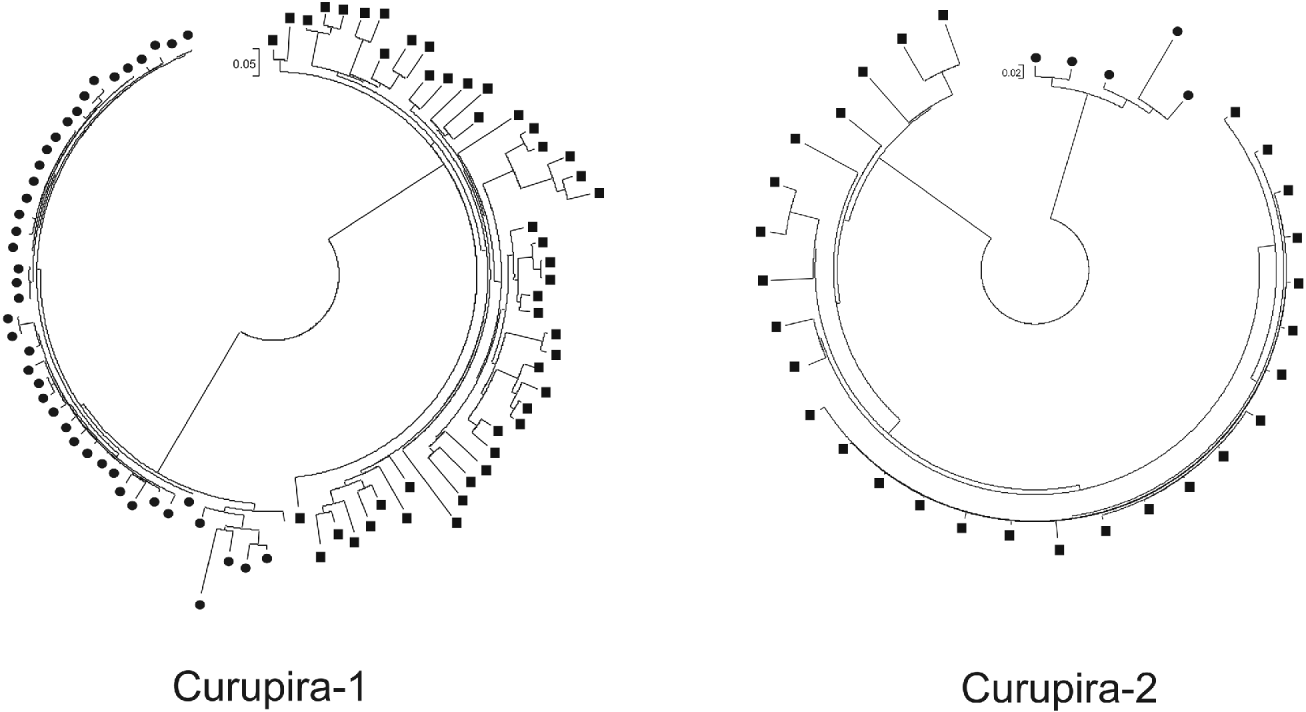
Fig. 5. Phylogenetic tree of Curupira-1 or -2 family members from Schistosoma mansoni and S. japonicum genomes. Trees were constructed using a separate multiple alignment for each Curupira element that included the different genomic copies of nucleotide sequences coding for the transposase domain in both S. mansoni (circles) and S. japonicum (squares) species and applying the approximately maximum likelihood algorithm.
DISCUSSION
In this work we described Curupira-1 and -2, two novel schistosome DNA transposons in the genomes of S. mansoni and S. japonicum. They have several distinct characteristics, such as the presence of a conserved MULE transposase domain (pfam10551) and the presence of 9 bp TSDs, which permitted us to classify them as members of the Mutator superfamily. Recently, a family of transposons named Phantom was the first from the Mutator superfamily to be described in a metazoan (Marquez & Pritham, Reference Marquez and Pritham2010). Here we show that a different lineage of Mutator transposons has also colonized the genomes of early metazoans. Curupira transposons are markedly different from Phantom transposons with respect to the size of TSD (9 bp in Curupira and 10 bp in Phantom), presence of TIRs (absent in Curupira and present in Phantom) and the presence of a SWIM zinc-finger domain in Curupira, but not in Phantom. In addition, phylogenetic analysis of the transposase domain suggests a very distant relationship between those two groups. This appears to confirm the proposal of two independent lineages colonizing the genomes of metazoan previously made by Hua-Van and Capy (Reference Hua-Van and Capy2008), based only on the analysis of a DDE region sampled from the genomes of different organisms.
Phylogenetic analysis suggests that SmCurupira-1 experienced a recent burst of transposition, similar to that verified for some major retrotransposon families of S. mansoni (Venancio et al. Reference Venancio, Wilson, Verjovski-Almeida and DeMarco2010). The presence of intact TSDs in several of the examined copies provides further evidence for recent transposition. Despite this recent expansion, we have been unable to identify a large amount of spliced transcripts for this element, as production of intact active copies would be expected. Instead, most of the transcripts, either sampled in the EST database or amplified by us using RT-PCR, were unspliced and appeared to be transcribed from truncated copies. It is possible that most of the intact copies are silenced and transcripts of truncated copies are produced by an alternate promoter, or are located in regions of the genome less prone to silencing. SmCurupira-2 had approximately the same estimated genome abundance as SjCurupira-1. However, only 5 copies with an intact transposase domain were sampled while SjCurupira-1 had 51 copies with this characteristic. In fact, most copies of SmCurupira-2 examined, and all those with intact extremities, lacked the transposase domain. The presence of TSDs in 4 out of 5 of the copies with intact extremities suggests a recent transposition of these elements. Taken together these data suggest that non-autonomous elements of SmCurupira-2 have recently been able to transpose using a transposase produced by another element. Considering that the S. mansoni genome sequence is incomplete, it is difficult to evaluate whether there is an intact SmCurupira-2 element that could act in trans or if this role is performed by an autonomous element from another family. In maize, the non-autonomous copies have been shown to outnumber the autonomous MuDR elements and are responsible for most of the mutations in mutator lines (Lisch, Reference Lisch2002). Therefore, the strategy of non-autonomous copies ensuring their transposition by the use of a much less numerous autonomous line appears to be recurrent in this class of elements.
Description of these novel schistosome transposable elements provides new insights into the schistosome genome dynamics and evolution. In addition, the presence of spliced transcripts and the integrity of some of the inspected copies suggest that they may represent active elements. If the latter suggestion is true, these elements could be potential tools to be used for the integration of specific genes into the parasite genome, leading to transgenic schistosomes and/or to permanently dividing schistosome cell lines, which may contribute significantly to future molecular studies of these parasites. However, further studies are warranted to confirm the production of an active transposase protein, as well as to determine the dynamics of interaction between these elements and the parasite genome before they can be seriously considered for this purpose.
FINANCIAL SUPPORT
This work was financed by a ‘Young Researcher’ grant from Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) to R.dM. and by the program ‘Institutos Nacionais de Ciência e Tecnologia’ (INCT) from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/MCT) and FAPESP.