Introduction
Major depressive disorder (MDD) is a common and disabling disorder with an age at onset most typically from late adolescence to middle adult life (Ferrari et al., Reference Ferrari, Charlson, Norman, Patten, Freedman, Murray, Vos and Whiteford2013). In multiple twin studies, lifetime MDD has been shown to have a heritability of approximately 40% with individual-specific environment contributing most of the remaining liability (Sullivan et al., Reference Sullivan, Neale and Kendler2000; Kendler et al., Reference Kendler, Gatz, Gardner and Pedersen2006). Polygenic studies have estimated that the sum of measured genetic variation explains 6–32% of the variance (SNP-h2) in risk of MDD (Lubke et al., Reference Lubke, Hottenga, Walters, Laurin, de Geus, Willemsen, Smit, Middeldorp, Penninx, Vink and Boomsma2012; Lee et al., Reference Lee, Ripke, Neale, Faraone, Purcell, Perlis, Mowry, Thapar, Goddard, Witte, Absher, Agartz, Akil, Amin, Andreassen, Anjorin, Anney, Anttila, Arking, Asherson, Azevedo, Backlund, Badner, Bailey, Banaschewski, Barchas, Barnes, Barrett, Bass, Battaglia, Bauer, Bayes, Bellivier, Bergen, Berrettini, Betancur, Bettecken, Biederman, Binder, Black, Blackwood, Bloss, Boehnke, Boomsma, Breen, Breuer, Bruggeman, Cormican, Buccola, Buitelaar, Bunney, Buxbaum, Byerley, Byrne, Caesar, Cahn, Cantor, Casas, Chakravarti, Chambert, Choudhury, Cichon, Cloninger, Collier, Cook, Coon, Cormand, Corvin, Coryell, Craig, Craig, Crosbie, Cuccaro, Curtis, Czamara, Datta, Dawson, Day, De Geus, Degenhardt, Djurovic, Donohoe, Doyle, Duan, Dudbridge, Duketis, Ebstein, Edenberg, Elia, Ennis, Etain, Fanous, Farmer, Ferrier, Flickinger, Fombonne, Foroud, Frank, Franke, Fraser, Freedman, Freimer, Freitag, Friedl, Frisen, Gallagher, Gejman, Georgieva, Gershon, Geschwind, Giegling, Gill, Gordon, Gordon-Smith, Green, Greenwood, Grice, Gross, Grozeva, Guan, Gurling, De Haan, Haines, Hakonarson, Hallmayer, Hamilton, Hamshere, Hansen, Hartmann, Hautzinger, Heath, Henders, Herms, Hickie, Hipolito, Hoefels, Holmans, Holsboer, Hoogendijk, Hottenga, Hultman, Hus, Ingason, Ising, Jamain, Jones, Jones, Jones, Tzeng, Kahler, Kahn, Kandaswamy, Keller, Kennedy, Kenny, Kent, Kim, Kirov, Klauck, Klei, Knowles, Kohli, Koller, Konte, Korszun, Krabbendam, Krasucki, Kuntsi, Kwan, Landen, Langstrom, Lathrop, Lawrence, Lawson, Leboyer, Ledbetter, Lee, Lencz, Lesch, Levinson, Lewis, Li, Lichtenstein, Lieberman, Lin, Linszen, Liu, Lohoff, Loo, Lord, Lowe, Lucae, MacIntyre, Madden, Maestrini, Magnusson, Mahon, Maier, Malhotra, Mane, Martin, Martin, Mattheisen, Matthews, Mattingsdal, McCarroll, McGhee, McGough, McGrath, McGuffin, McInnis, McIntosh, McKinney, McLean, McMahon, McMahon, McQuillin, Medeiros, Medland, Meier, Melle, Meng, Meyer, Middeldorp, Middleton, Milanova, Miranda, Monaco, Montgomery, Moran, Moreno-De-Luca, Morken, Morris, Morrow, Moskvina, Muglia, Muhleisen, Muir, Muller-Myhsok, Murtha, Myers, Myin-Germeys, Neale, Nelson, Nievergelt, Nikolov, Nimgaonkar, Nolen, Nothen, Nurnberger, Nwulia, Nyholt, O'Dushlaine, Oades, Olincy, Oliveira, Olsen, Ophoff, Osby, Owen, Palotie, Parr, Paterson, Pato, Pato, Penninx, Pergadia, Pericak-Vance, Pickard, Pimm, Piven, Posthuma, Potash, Poustka, Propping, Puri, Quested, Quinn, Ramos-Quiroga, Rasmussen, Raychaudhuri, Rehnstrom, Reif, Ribases, Rice, Rietschel, Roeder, Roeyers, Rossin, Rothenberger, Rouleau, Ruderfer, Rujescu, Sanders, Sanders, Santangelo, Sergeant, Schachar, Schalling, Schatzberg, Scheftner, Schellenberg, Scherer, Schork, Schulze, Schumacher, Schwarz, Scolnick, Scott, Shi, Shilling, Shyn, Silverman, Slager, Smalley, Smit, Smith, Sonuga-Barke, St Clair, State, Steffens, Steinhausen, Strauss, Strohmaier, Stroup, Sutcliffe, Szatmari, Szelinger, Thirumalai, Thompson, Todorov, Tozzi, Treutlein, Uhr, van den Oord, Van Grootheest, Van Os, Vicente, Vieland, Vincent, Visscher, Walsh, Wassink, Watson, Weissman, Werge, Wienker, Wijsman, Willemsen, Williams, Willsey, Witt, Xu, Young, Yu, Zammit, Zandi, Zhang, Zitman, Zollner, Devlin, Kelsoe, Sklar, Daly, O'Donovan, Craddock, Sullivan, Smoller, Kendler, Wray, Genomi and Genetic2013; Hyde et al., Reference Hyde, Nagle, Tian, Chen, Paciga, Wendland, Tung, Hinds, Perlis and Winslow2016; Direk et al., Reference Direk, Williams, Smith, Ripke, Air, Amare, Amin, Baune, Bennett, Blackwood, Boomsma, Breen, Buttenschon, Byrne, Borglum, Castelao, Cichon, Clarke, Cornelis, Dannlowski, De Jager, Demirkan, Domenici, van Duijn, Dunn, Eriksson, Esko, Faul, Ferrucci, Fornage, de Geus, Gill, Gordon, Grabe, van Grootheest, Hamilton, Hartman, Heath, Hek, Hofman, Homuth, Horn, Jan Hottenga, Kardia, Kloiber, Koenen, Kutalik, Ladwig, Lahti, Levinson, Lewis, Lewis, Li, Llewellyn, Lucae, Lunetta, MacIntyre, Madden, Martin, McIntosh, Metspalu, Milaneschi, Montgomery, Mors, Mosley, Murabito, Muller-Myhsok, Nothen, Nyholt, O'Donovan, Penninx, Pergadia, Perlis, Potash, Preisig, Purcell, Quiroz, Raikkonen, Rice, Rietschel, Rivera, Schulze, Shi, Shyn, Sinnamon, Smit, Smoller, Snieder, Tanaka, Tansey, Teumer, Uher, Umbricht, Van der Auwera, Ware, Weir, Weissman, Willemsen, Yang, Zhao, Tiemeier and Sullivan2017; Wray et al., Reference Wray, Ripke, Mattheisen, Trzaskowski, Byrne, Abdellaoui, Adams, Agerbo, Air, Andlauer, Bacanu, Baekvad-Hansen, Beekman, Bigdeli, Binder, Blackwood, Bryois, Buttenschon, Bybjerg-Grauholm, Cai, Castelao, Christensen, Clarke, Coleman, Colodro-Conde, Couvy-Duchesne, Craddock, Crawford, Crowley, Dashti, Davies, Deary, Degenhardt, Derks, Direk, Dolan, Dunn, Eley, Eriksson, Escott-Price, Kiadeh, Finucane, Forstner, Frank, Gaspar, Gill, Giusti-Rodriguez, Goes, Gordon, Grove, Hall, Hannon, Hansen, Hansen, Herms, Hickie, Hoffmann, Homuth, Horn, Hottenga, Hougaard, Hu, Hyde, Ising, Jansen, Jin, Jorgenson, Knowles, Kohane, Kraft, Kretzschmar, Krogh, Kutalik, Lane, Li, Li, Lind, Liu, Lu, MacIntyre, MacKinnon, Maier, Maier, Marchini, Mbarek, McGrath, McGuffin, Medland, Mehta, Middeldorp, Mihailov, Milaneschi, Milani, Mill, Mondimore, Montgomery, Mostafavi, Mullins, Nauck, Ng, Nivard, Nyholt, O'Reilly, Oskarsson, Owen, Painter, Pedersen, Pedersen, Peterson, Pettersson, Peyrot, Pistis, Posthuma, Purcell, Quiroz, Qvist, Rice, Riley, Rivera, Saeed Mirza, Saxena, Schoevers, Schulte, Shen, Shi, Shyn, Sigurdsson, Sinnamon, Smit, Smith, Stefansson, Steinberg, Stockmeier, Streit, Strohmaier, Tansey, Teismann, Teumer, Thompson, Thomson, Thorgeirsson, Tian, Traylor, Treutlein, Trubetskoy, Uitterlinden, Umbricht, Van der Auwera, van Hemert, Viktorin, Visscher, Wang, Webb, Weinsheimer, Wellmann, Willemsen, Witt, Wu, Xi, Yang, Zhang, Arolt, Baune, Berger, Boomsma, Cichon, Dannlowski, de Geus, DePaulo, Domenici, Domschke, Esko, Grabe, Hamilton, Hayward, Heath, Hinds, Kendler, Kloiber, Lewis, Li, Lucae, Madden, Magnusson, Martin, McIntosh, Metspalu, Mors, Mortensen, Muller-Myhsok, Nordentoft, Nothen, O'Donovan, Paciga, Pedersen, Penninx, Perlis, Porteous, Potash, Preisig, Rietschel, Schaefer, Schulze, Smoller, Stefansson, Tiemeier, Uher, Volzke, Weissman, Werge, Winslow, Lewis, Levinson, Breen, Borglum and Sullivan2018). However, most of the genetic risk has not been linked to specific polymorphisms (Ripke et al., Reference Ripke, Wray, Lewis, Hamilton, Weissman, Breen, Byrne, Blackwood, Boomsma, Cichon, Heath, Holsboer, Lucae, Madden, Martin, McGuffin, Muglia, Noethen, Penninx, Pergadia, Potash, Rietschel, Lin, Muller-Myhsok, Shi, Steinberg, Grabe, Lichtenstein, Magnusson, Perlis, Preisig, Smoller, Stefansson, Uher, Kutalik, Tansey, Teumer, Viktorin, Barnes, Bettecken, Binder, Breuer, Castro, Churchill, Coryell, Craddock, Craig, Czamara, De Geus, Degenhardt, Farmer, Fava, Frank, Gainer, Gallagher, Gordon, Goryachev, Gross, Guipponi, Henders, Herms, Hickie, Hoefels, Hoogendijk, Hottenga, Iosifescu, Ising, Jones, Jones, Jung-Ying, Knowles, Kohane, Kohli, Korszun, Landen, Lawson, Lewis, Macintyre, Maier, Mattheisen, McGrath, McIntosh, McLean, Middeldorp, Middleton, Montgomery, Murphy, Nauck, Nolen, Nyholt, O'Donovan, Oskarsson, Pedersen, Scheftner, Schulz, Schulze, Shyn, Sigurdsson, Slager, Smit, Stefansson, Steffens, Thorgeirsson, Tozzi, Treutlein, Uhr, van den Oord, Van Grootheest, Volzke, Weilburg, Willemsen, Zitman, Neale, Daly, Levinson and Sullivan2013; Converge consortium, 2015; Geschwind and Flint, Reference Geschwind and Flint2015; Van der Auwera et al., Reference Van der Auwera, Peyrot, Milaneschi, Hertel, Baune, Breen, Byrne, Dunn, Fisher, Homuth, Levinson, Lewis, Mills, Mullins, Nauck, Pistis, Preisig, Rietschel, Ripke, Sullivan, Teumer, Volzke, Boomsma, Wray, Penninx and Grabe2018). One of the several factors contributing to this discrepancy could be age-related variation in risk factors (Korten et al., Reference Korten, Comijs, Lamers and Penninx2012; Power et al., Reference Power, Tansey, Buttenschon, Cohen-Woods, Bigdeli, Hall, Kutalik, Lee, Ripke, Steinberg, Teumer, Viktorin, Wray, Arolt, Baune, Boomsma, Borglum, Byrne, Castelao, Craddock, Craig, Dannlowski, Deary, Degenhardt, Forstner, Gordon, Grabe, Grove, Hamilton, Hayward, Heath, Hocking, Homuth, Hottenga, Kloiber, Krogh, Landen, Lang, Levinson, Lichtenstein, Lucae, MacIntyre, Madden, Magnusson, Martin, McIntosh, Middeldorp, Milaneschi, Montgomery, Mors, Muller-Myhsok, Nyholt, Oskarsson, Owen, Padmanabhan, Penninx, Pergadia, Porteous, Potash, Preisig, Rivera, Shi, Shyn, Sigurdsson, Smit, Smith, Stefansson, Stefansson, Strohmaier, Sullivan, Thomson, Thorgeirsson, Van der Auwera, Weissman, Breen and Lewis2017). Results from studies using diagnostic interviews of twins indicate completely stable genetic risk factors for MDD from the 20s to 30s (Torvik et al., Reference Torvik, Rosenstrom, Ystrom, Tambs, Roysamb, Czajkowski, Gillespie, Knudsen, Kendler and Reichborn-Kjennerud2017), and in MDD assessed two times 1.5 years apart (Kendler et al., Reference Kendler, Neale, Kessler, Heath and Eaves1993), and four times over a decade in adulthood (Kendler and Gardner, Reference Kendler and Gardner2017). Studies on symptoms of depression and/or anxiety have found small or no changes in genetic risk factors during adulthood (Gillespie et al., Reference Gillespie, Kirk, Evans, Heath, Hickie and Martin2004; Cerda et al., Reference Cerda, Sagdeo, Johnson and Galea2010; Nivard et al., Reference Nivard, Dolan, Kendler, Kan, Willemsen, van Beijsterveldt, Lindauer, van Beek, Geels, Bartels, Middeldorp and Boomsma2015), but there seem to be genetic factors specific to childhood and adolescence (Kendler et al., Reference Kendler, Gardner and Lichtenstein2008; Nivard et al., Reference Nivard, Dolan, Kendler, Kan, Willemsen, van Beijsterveldt, Lindauer, van Beek, Geels, Bartels, Middeldorp and Boomsma2015; Waszczuk et al., Reference Waszczuk, Zavos, Gregory and Eley2016) and old age (Gillespie et al., Reference Gillespie, Kirk, Evans, Heath, Hickie and Martin2004; Petkus et al., Reference Petkus, Gatz, Reynolds, Kremen and Wetherell2016).
Conflicting information exists about the temporal stability of the environmental risk factors for MDD. One view is that the effects of such risk factors rapidly decrease over time, disappearing in as short a time period as a single year (Kendler et al., Reference Kendler, Neale, Kessler, Heath and Eaves1993; Dunn et al., Reference Dunn, Brown, Dai, Rosand, Nugent, Amstadter and Smoller2015), and that the environment is therefore not responsible for the longer-term stability of risk. In this view, the stability of MDD is entirely due to genetic factors, whereas environmental events produce variation around this ‘set point’. By contrast, a range of studies shows that early severe adversities such as childhood sexual abuse can have enduring effects on the risk of MDD for decades (Hammen, Reference Hammen2005). Most such studies are genetically uninformative and therefore unable to determine to what extent the environment contributes to stability. The major findings from twin studies concerning this has indicated no (Kendler et al., Reference Kendler, Neale, Kessler, Heath and Eaves1993; Torvik et al., Reference Torvik, Rosenstrom, Ystrom, Tambs, Roysamb, Czajkowski, Gillespie, Knudsen, Kendler and Reichborn-Kjennerud2017) or low (Kendler and Gardner, Reference Kendler and Gardner2010, Reference Kendler and Gardner2017) stability in environmental causes of MDD and symptoms of anxiety and depression in adulthood (Gillespie et al., Reference Gillespie, Kirk, Evans, Heath, Hickie and Martin2004; Nivard et al., Reference Nivard, Dolan, Kendler, Kan, Willemsen, van Beijsterveldt, Lindauer, van Beek, Geels, Bartels, Middeldorp and Boomsma2015; Waszczuk et al., Reference Waszczuk, Zavos, Gregory and Eley2016). These studies rely on self-reported symptoms, which include measurement error that can lead to underestimates of environmental stability. In addition, studies with a long duration between follow-ups were not able to study short-term stability. The most informative study to date on this question (Kendler and Gardner, Reference Kendler and Gardner2017) suggests that about 17% of the environmental influences on MDD in the last year in are stable over 8 years and the remainder is occasion-specific. Both clinical and molecular genetic work would benefit from a better understanding of the degree of stability of the genetic and environmental risk factors for MDD. This can be achieved if MDD is observed over a long-time window with assessments close in time.
The purpose of this study is to examine to what degree genetic and environmental influences on clinically assessed MDD are stable between age 18 and 45 by using a population-based twin sample with continuously updated registry data from primary care.
Methods
Sample
The data consist of registry-based information on 11 727 Norwegian twins born between 1967 and 1991 who were recorded in the Norwegian Twin Registry. In total, 21 517 twins identified through the mandatory Norwegian Medical Birth Registry were invited to be part of the twin registry. Among these, 433 (2.0%) had an unknown address, whereas 11 608 (53.9%) gave consent. In addition, 116 twins consented to registry linking without being permanent members of the registry, and three twins born abroad self-recruited. Individuals with possible schizophrenia or bipolar disorder (n = 163) were excluded from the analyses. The analyzed sample thus consisted of 11 564 individuals (59.4% women). Zygosity was determined by a combination of questionnaire items and genotyping of a subsample. There were 1860 complete MZ and 2190 complete DZ twin pairs as well as 3445 single twins with known zygosity. Using unique person-identification numbers assigned at birth, we linked the twin registry to demographic registries and treatment data from governmentally funded primary care for the years 2006–2015. As consent was gathered in 2016, there was no attrition. The twins were on average 27.7 years old at the beginning of 2006 (range 14–38), and 37.7 at the end of 2015.
Ethics
The study was approved by the Regional Ethical Committee for Medical and Health Research Ethics, and written informed consent was obtained from all participants.
Measures
Primary care data
All individuals who legally reside in Norway are members of the National Insurance Scheme and assigned a general practitioner. General practitioners and other health service providers, such as emergency rooms, send billing information to The Norwegian Health Economics Administration (Helfo) along with a diagnosis or reason for the visit in order to receive reimbursements. Due to economic incentives, it is unlikely that health visits go unreported. Diagnostic information is coded according to the International Classification of Primary Care (ICPC-2) (World Organization of National Colleges Academies, 2005) and registered in the database Control and Payment of Health Reimbursements operated by the Norwegian Directorate of Health. The ICPC-2 contains both diagnoses and complaints. In this study, we analyze visits registered with the diagnosis ‘P76 – Depressive disorder’ as MDD. We have previously demonstrated that this diagnosis is strongly phenotypically and nearly fully genetically correlated both with diagnoses given in specialist care (F32 and F33) and with diagnoses from structured interviews (Torvik et al., Reference Torvik, Ystrom, Gustavson, Rosenstrom, Bramness, Gillespie, Aggen, Kendler and Reichborn-Kjennerud2018). Being registered at least once with either ‘P72 – Schizophrenia’ or ‘P73 Affective disorder’ (n = 163) was used as an exclusion criterion.
Demographic data
The data were linked to demographic information on educational attainment from The Norwegian Educational Database and information on income and marital status from The Tax Database, both databases operated by Statistics Norway. At the end of the observational period (in 2015), 18.1% had master's degree or equivalent, 40.4% had bachelor's degree or equivalent, 33.5% had completed high school, and 8.0% had primary education only.
Statistical analyses
We first described the associations of MDD with sex, age and educational attainment in multiple logistic regression models, and then tested the association with income, marriage and divorce adjusted for these variables. We did this in order to describe the sample and to test whether MDD measured in the registries related to known characteristics of individuals with MDD.
We applied an accelerated longitudinal twin design to study the development of depression from ages 18 to 45. In this design, each individual is followed for a limited amount of time, here 10 years, and where variation in individuals’ age across the sample permits an examination of development over a longer period. In the current analyses, we analyzed the occurrence of MDD in 2-year windows from ages 18 to 45. As shown in online Supplementary Table S1, this resulted in 14-time intervals which were scored ‘0’ if there were no MDD entries in the registry for that period or ‘1’ if there were one or more MDD entries. We did not model MDD prior to age 18 or above age 45, due to the low number of observations and differences relating to the organization of child mental health services.
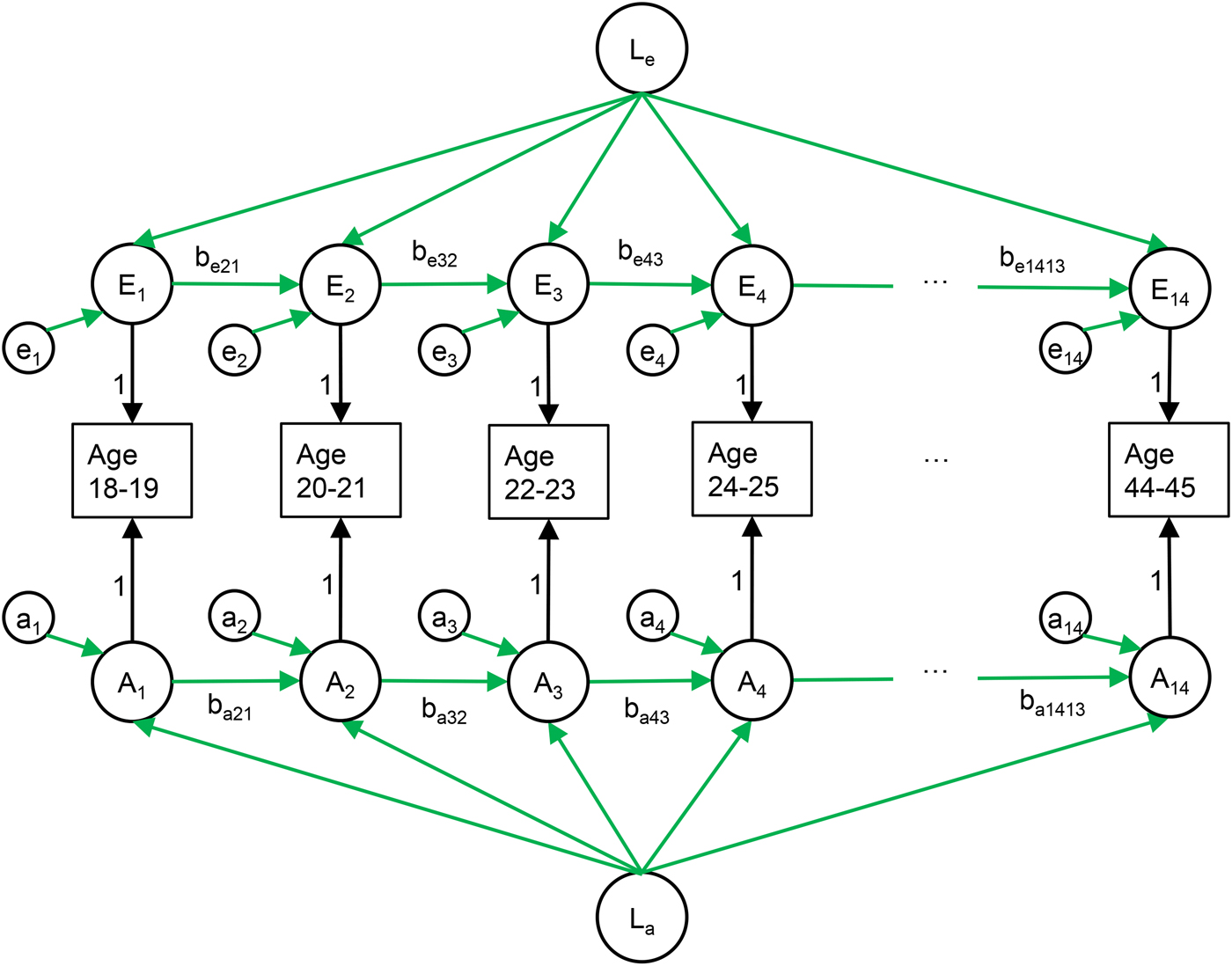
We modelled the genetic and environmental sources of individual differences in risk of MDD within and across time by using multivariate twin analyses for binary data with different prevalences (thresholds) for men and women at each age. Monozygotic (MZ) twins share all their genes and dizygotic (DZ) twins share on average half of the genes that vary in the population. Utilizing this difference, stability and change in depression can be ascribed to varying combinations of additive genetic factors (A), shared environmental factors (C), and individual-specific or non-shared environmental factors, which includes measurement error (E). For illustration, we consider a twin pair where one member has MDD. If the stability between time-points is due to E factors alone, the depressed twin will have an elevated risk of MDD at the next observation, but not the co-twin. If the stability is due to C factors alone, both twins, regardless of their genetic relatedness, will have the same elevated risk of future MDD as the initially depressed twin, and this is true for MZ and DZ twin pairs alike. If, however, the stability is due to A factors alone, MZ co-twins are equally likely to be depressed at the next point in time, whereas DZ co-twins will have a less elevated risk due to sharing only half their genes.
We used the Cholesky decomposition to freely estimates of the correlations between genetic influences on MDD at the different ages, and similarly for the environmental influences (Neale and Cardon, Reference Neale and Cardon1992). We then applied a model that includes two processes: (i) stable components of A, C, and E that influence all time points; and (ii) auto-regressive components of A, C, and E, which make each observation in part dependent on the genetic and environmental factors active at the previous observation plus new variation. Thus, we can separate enduring individual set-point from temporary stability in each of the three biometric components. See Fig. 1 for an illustration of the model and the Figure legend for a more detailed explanation. We compared this model to the Cholesky decomposition to test if it adequately represented the data. Simpler, more restricted variants of the model were then tested by removing specific paths from the model or setting several paths to equal. We restricted paths between adjacent time points to be equal in order to test whether the stability of MDD varied between life-phases. We then tested the presence of new genetic or shared environmental influences during the observational period by setting the effects of these to zero, and tested whether there were any auto-regression by setting the genetic and environmental path between adjacent time-points to zero. Finally, we tested the risk factors by setting these to zero. The models were fitted to raw, ordinal data using the OpenMx 2.7.16 package for R. The raw data method utilizes all data, from both complete and incomplete pairs, and allows estimating effects for the full age range, although each individual is observed for only 10 years. We used a threshold-liability model, which models ordinal categories as arising from estimated thresholds on an underlying normal distribution (Falconer, Reference Falconer1965). The twins in incomplete pairs are useful in estimating stability and change but do not contribute towards the estimation of genetic and environmental factors. We determined the goodness of fit using likelihood ratio chi-square tests and by comparing the sample-size adjusted Bayesian information criterion (sBIC). By the principle of parsimony, models with the lowest sBIC were preferred (Sclove, Reference Sclove1987).
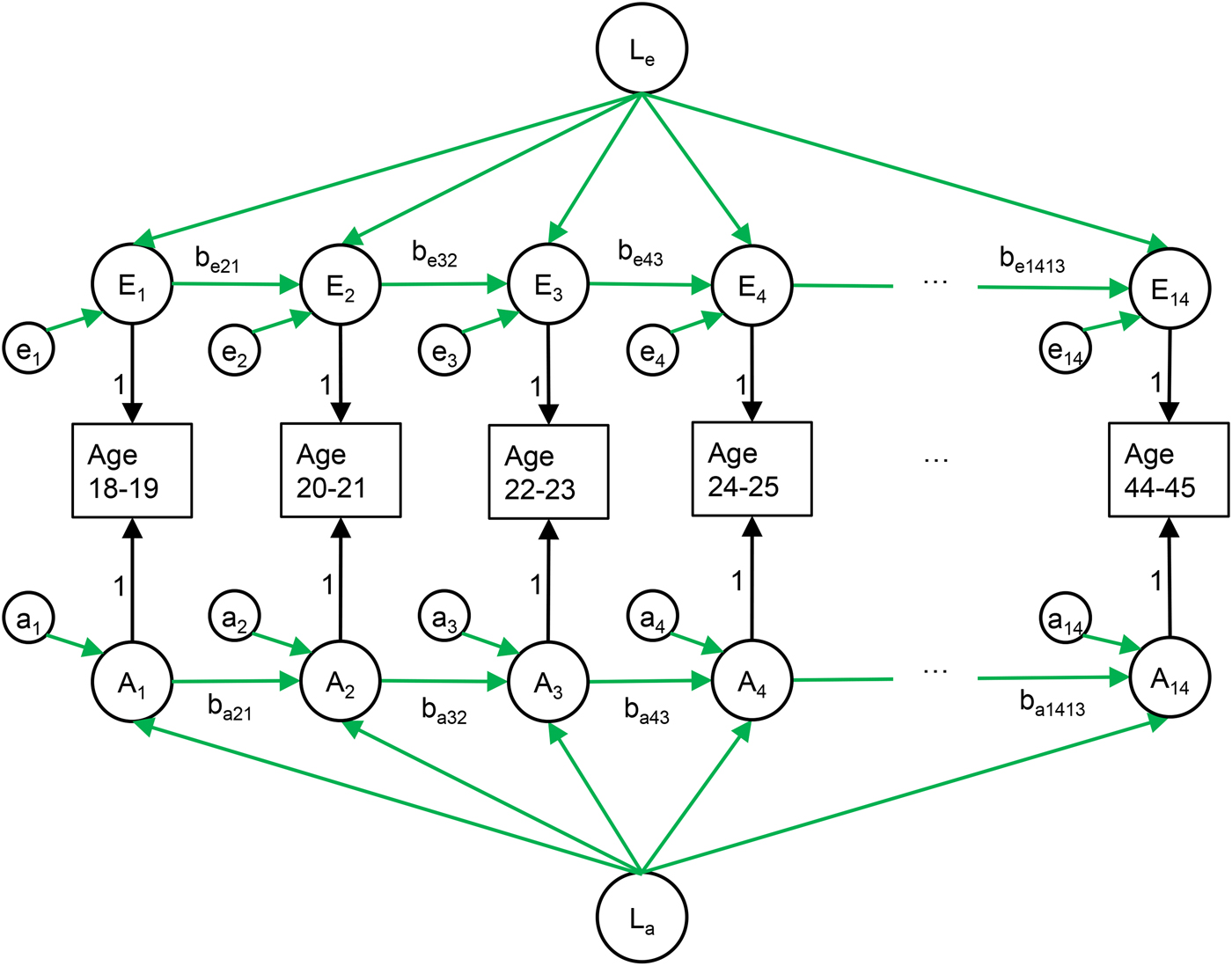
Fig. 1. The longitudinal model of major depressive disorder (MDD) in primary care from age 18 to 45 in 2-year prevalence windows. The environmental variation in risk of MDD (upper part) consists of three parts: (i) a latent factor common to all time points (Le), (ii) new variation (et), and (iii) effects from previous time points transmitted via the auto-regression (bet,t−1). The genetic variation in risk of MDD (lower part) has the same structure. Parallel structures were also modelled for shared environmental influences, for simplicity not shown in this figure.
Results
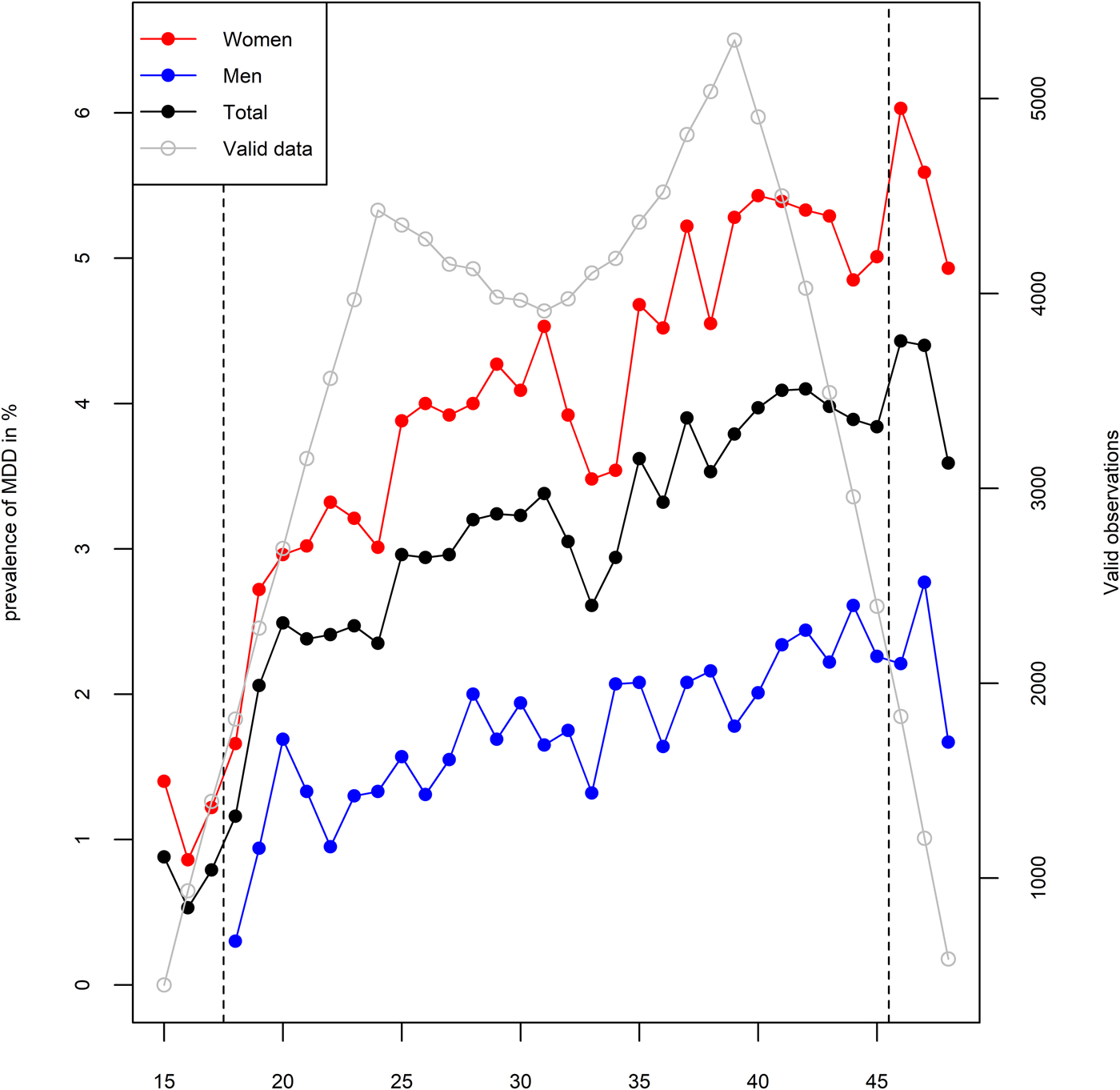
In an average year, 1.8% of men and 4.2% of women were registered at least once with MDD, although as depicted in Fig. 2, this varied by age. During the observational period of 10 years, 366 men (7.8%) and 1210 women (17.6%) were registered with at least one episode of MDD. We ran a series of multiple logistic regression analyses in order to test the associations between MDD and demographic characteristics. All of these analyses are adjusted for age, sex, and educational attainment. MDD was more common among women with an odds ratio (OR) of 2.71 (95% CI 2.39–3.07), individuals with higher age with an OR of 1.02 (95% CI 1.01–1.02) per year, and less common among individuals with higher educational attainment with an OR of 0.63 (95% CI 0.59–0.67) per level of education. Being registered at least once with MDD was associated with an annual income loss of 75 000 Norwegian kroner (95% CI 63 000–88 000) at the end of the observational period, which corresponds to 16.8% of the median income in the sample. MDD was also associated with a lower probability of being ever married (OR = 0.76, 95% CI 0.67–0.86) and a higher probability of divorce among those who married (OR = 2.52, 95% CI 2.07–3.06). Year of birth was not statistically significantly associated with MDD after adjustment for age and sex (OR = 1.02, 95% CI 0.99–1.05). A demographic breakdown of the sample by zygosity is provided in online Supplementary Table S2.
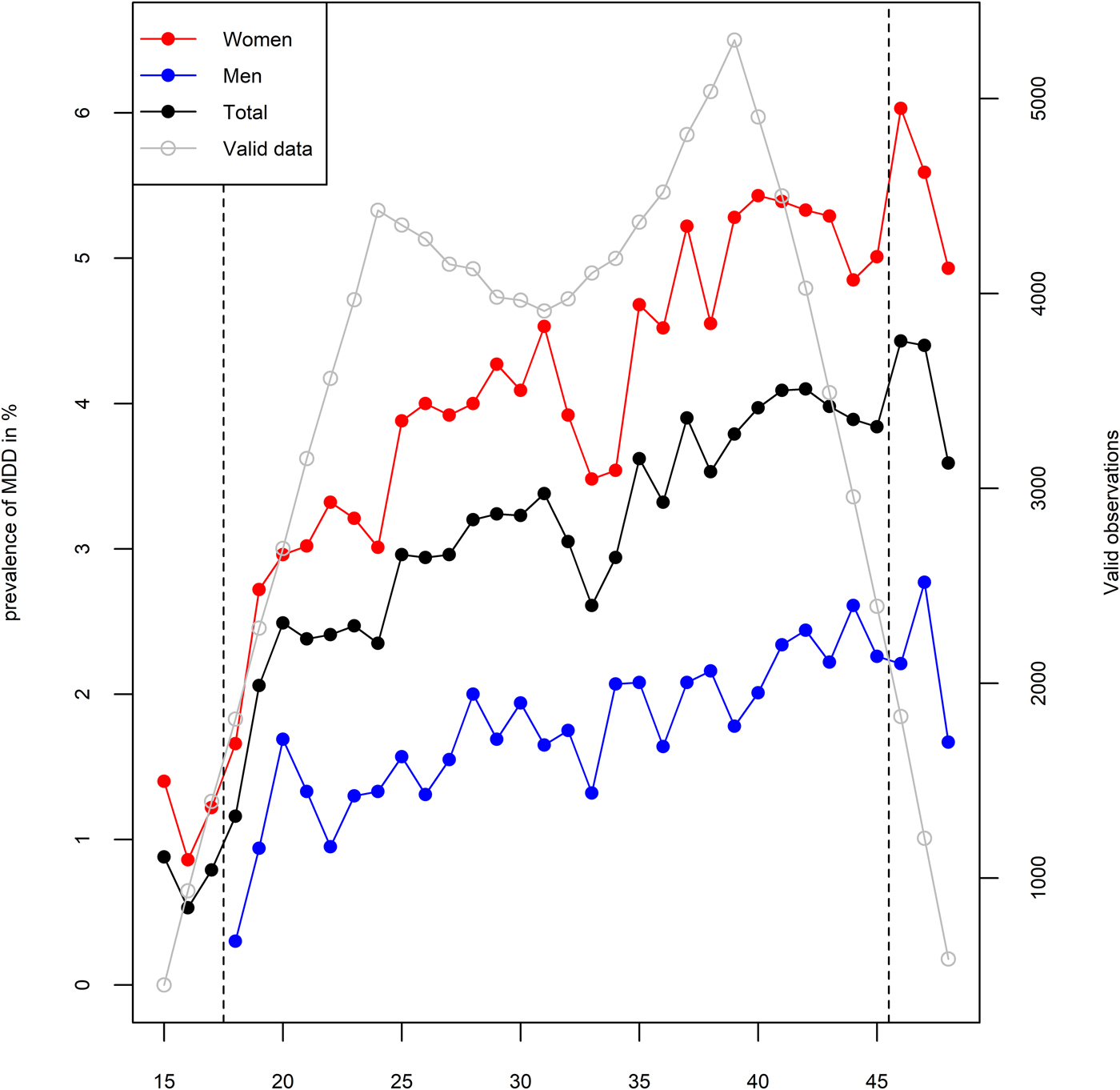
Fig. 2. One-year prevalence of major depressive disorder (MDD) in primary care among women (red), men (blue), and total (black) in %, by age. Grey line represents the relative amount of available data at each age.
The analyses of stability and change were based on 2-year prevalence windows. The average phenotypic tetrachoric correlation of registered MDD between adjacent 2-year prevalence windows was +0.75. Correspondingly, over 4, 6 and 8 years, the average correlation was respectively +0.60, +0.47, and +0.47. Thus, observations close in time have higher correlations than distant observations, but after some time, they seem to stabilize. A full phenotypic correlation matrix is provided in online Supplementary Table S3.
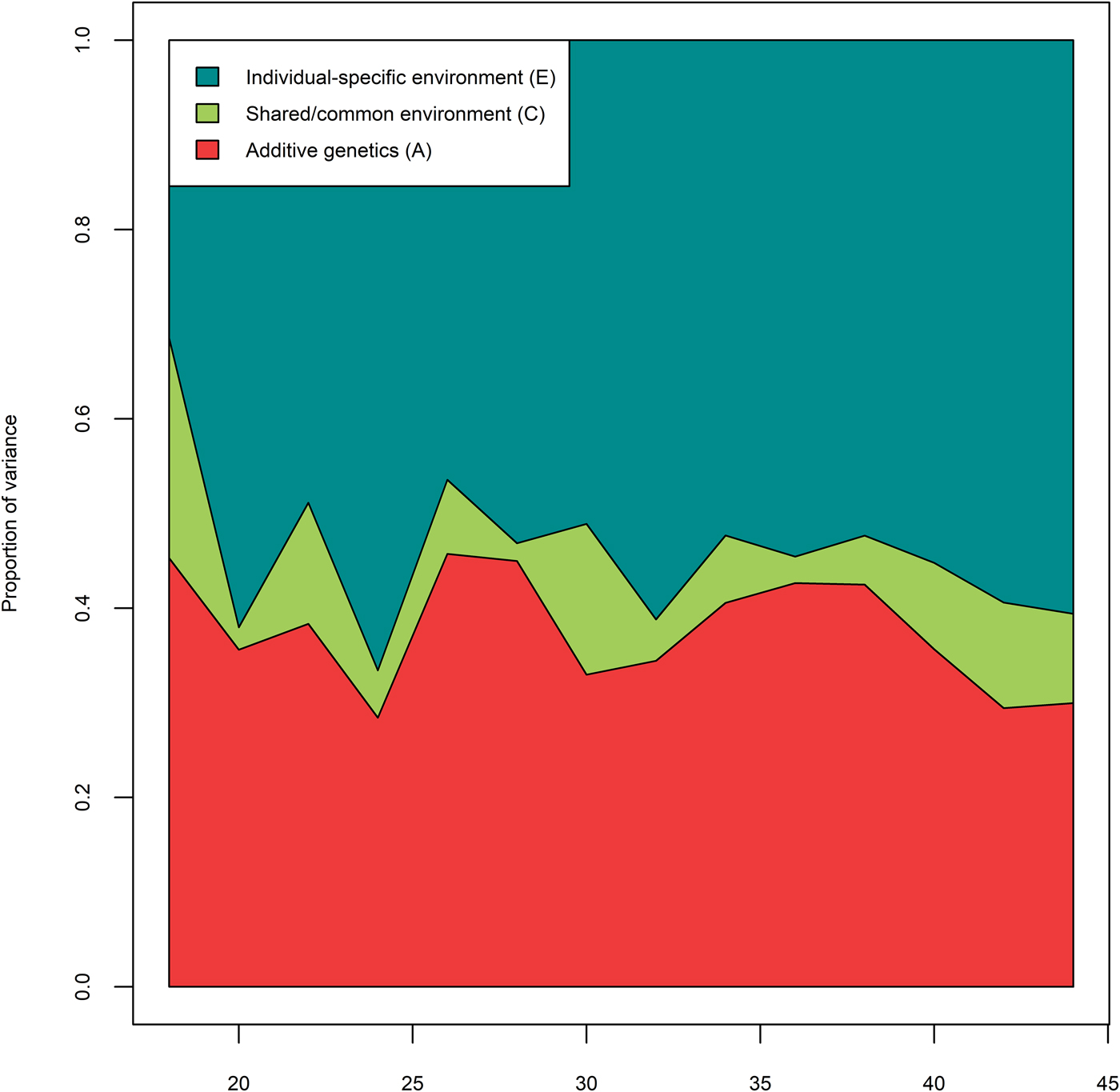
We first applied an unrestricted full correlational model (Cholesky decomposition) to estimate freely how A, C, and E contributed to MDD at each 2-year prevalence window and the correlations between MDD across age. Figure 3 shows the proportion of variance explained by A, C, and E factors in each 2-year prevalence window. Averaged across all ages, genetic factors (A) accounted for 37.5% of the variation in MDD, shared environmental (C) factors for 8.4%, and individual-specific (E) environmental factors for 54.1%. All fit indices for the biometric modelling is provided in online Supplementary Table S4. Compared with the fully saturated Cholesky, the longitudinal model (Fig. 1) had a better fit in terms of sBIC (ΔsBIC = −1314.55). We tested whether MDD was more stable in some life-phases than in others by testing if the paths between adjacent time points could be set to be constant across age for A, C and E, instead of estimating each path separately. This improved the model parsimony (ΔsBIC = −185.23). Next, we tested whether the genetic effects present at age 18 could explain the genetic risk at all subsequent observational windows, and similarly for shared environmental risk. A model without either novel genetic influences (‘genetic innovation’) or novel shared environmental effects provided the better fit (ΔsBIC = −129.18). We further tested if the influences of for A, C, and E were enduring and affected MDD at subsequent prevalence windows via the auto-regression. This process would describe A, C, or E effects that are still active over the next observational period, but not throughout the entire observational window. Such autoregressive models would be favored if influences on observations close in time were more strongly correlated than influences on distant observations. We found that removing the genetic effects between adjacent time points improved the model (ΔsBIC = −11.32), as did the removal of the shared-environmental effects between adjacent time points (ΔsBIC = −10.24). However, removing the individual-specific effects between adjacent time points caused model fit to deteriorate (ΔsBIC = + 219.62). In subsequent models, the individual-specific environment is dependent on previous observations, whereas additive genetic and shared environmental effects are stable throughout the observational period. Finally, we tested whether there were stable risk factors for A, C, and E by setting each of these to zero. A stable genetic risk factor could not be removed from the model (ΔsBIC = + 13.68), but the two stable environmental risk factors (C, E, and both) could be removed with a slight improvement in fit (ΔsBIC = −5.62, ΔsBIC = −5.73, and ΔsBIC = −11.48, respectively). This implies that there are no influences of shared environment present in the model and that the shared environmental influences in Fig. 3 are not significant.

Fig. 3. Relative contributions of genetic (A; red), shared environmental (C; green) and individual-specific environment (E; blue) to MDD in primary care by age. Results from Cholesky decomposition. As the data were binary, the variance is fixed to unity.
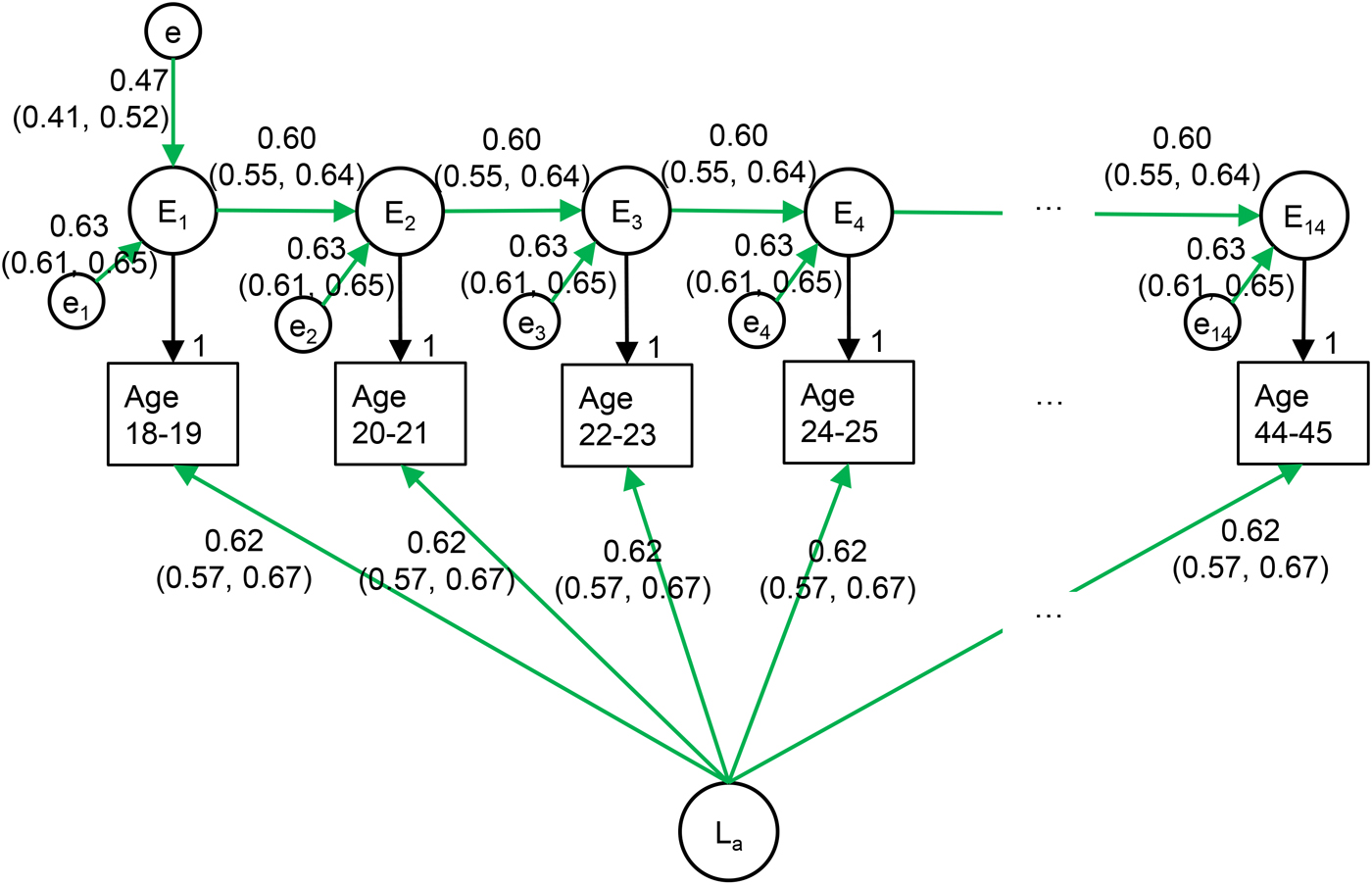
In the best fitting model, shown in Fig. 4, the genetic factors are stable across time, whereas the environment is individual-specific and changing at a constant rate. In this model, genetic factors explain 38.0% of the variance in MDD at each time-point and account for all long-term stability. Environmental factors correlate +0.60 over 2 years and +0.36 (0.602) over 4 years. New individual-specific environmental influences explain 39.5% of the variation in MDD at any given point in time, whereas 22.5% of the variance is due to environmental influences from earlier time-points.

Fig. 4. Best fitting longitudinal model of MDD in primary care.
Discussion
We examined a population-based twin sample with longitudinal information on clinically assessed depression and found that a simple developmental model best explained the genetic and environmental structure of clinically assessed MDD from age 18 to 45. The model entails three notable features: (i) complete stability of genetic risk factors, (ii) high stability of the individual-specific environment over short periods of time, but minimal long-term environmental stability, and (iii) no significant effects of shared environment.
We found stable genetic influences in MDD between ages 18 and 45. Although previous studies have not investigated genetic continuity in clinically assessed MDD over this long age span, the findings are consistent with previous research on MDD over shorter time-periods (Kendler et al., Reference Kendler, Neale, Kessler, Heath and Eaves1993; Kendler and Gardner, Reference Kendler and Gardner2017; Torvik et al., Reference Torvik, Rosenstrom, Ystrom, Tambs, Roysamb, Czajkowski, Gillespie, Knudsen, Kendler and Reichborn-Kjennerud2017), and with research on symptoms of anxiety and depression (Gillespie et al., Reference Gillespie, Kirk, Evans, Heath, Hickie and Martin2004; Cerda et al., Reference Cerda, Sagdeo, Johnson and Galea2010; Nivard et al., Reference Nivard, Dolan, Kendler, Kan, Willemsen, van Beijsterveldt, Lindauer, van Beek, Geels, Bartels, Middeldorp and Boomsma2015). This finding is important for molecular genetic studies of MDD because they suggest that there is no age-related heterogeneity from early to middle adulthood. One may, therefore, use heterogenous samples without worrying that they might be identifying distinct genetic risk variants acting at different ages. This is unlike for instance for alcohol use disorder, where changing genetic influences has been found during adulthood (Long et al., Reference Long, Lonn, Sundquist, Sundquist and Kendler2017; Torvik et al., Reference Torvik, Rosenstrom, Ystrom, Tambs, Roysamb, Czajkowski, Gillespie, Knudsen, Kendler and Reichborn-Kjennerud2017). There are, however, indications that the genetic effects for MDD could be different in childhood, early adolescence and old age (Gillespie et al., Reference Gillespie, Kirk, Evans, Heath, Hickie and Martin2004; Nivard et al., Reference Nivard, Dolan, Kendler, Kan, Willemsen, van Beijsterveldt, Lindauer, van Beek, Geels, Bartels, Middeldorp and Boomsma2015; Petkus et al., Reference Petkus, Gatz, Reynolds, Kremen and Wetherell2016; Waszczuk et al., Reference Waszczuk, Zavos, Gregory and Eley2016). Whereas we did not specifically study age at first onset, our results may seem to deviate from a molecular genetic study finding a locus associated with age at onset (Power et al., Reference Power, Tansey, Buttenschon, Cohen-Woods, Bigdeli, Hall, Kutalik, Lee, Ripke, Steinberg, Teumer, Viktorin, Wray, Arolt, Baune, Boomsma, Borglum, Byrne, Castelao, Craddock, Craig, Dannlowski, Deary, Degenhardt, Forstner, Gordon, Grabe, Grove, Hamilton, Hayward, Heath, Hocking, Homuth, Hottenga, Kloiber, Krogh, Landen, Lang, Levinson, Lichtenstein, Lucae, MacIntyre, Madden, Magnusson, Martin, McIntosh, Middeldorp, Milaneschi, Montgomery, Mors, Muller-Myhsok, Nyholt, Oskarsson, Owen, Padmanabhan, Penninx, Pergadia, Porteous, Potash, Preisig, Rivera, Shi, Shyn, Sigurdsson, Smit, Smith, Stefansson, Stefansson, Strohmaier, Sullivan, Thomson, Thorgeirsson, Van der Auwera, Weissman, Breen and Lewis2017). This potential discrepancy may be explained by our exclusion of individuals who developed schizophrenia or bipolar disorder, which were related to early onset MDD in the aforementioned study.
A fundamentally different mechanism emerged in the individual-specific environment, which had no stable component but rather was explained by a combination of previous plus new or emergent environmental risks. This implies that events that increase the risk for MDD at one point persist over time with their effects decreasing at an approximately the same rate throughout adulthood. Whereas the association is rather strong across short time-spans, and theoretically never fully disappears, it dissipates quickly, so that environmental factors relevant at one time point explain 62% of the variation at that time point, but only 10% of the total variation in MDD risk after 3.5 years, and only 1% after 8 years. These results are commensurate first with studies finding that depressive episodes predict future depressive episodes (Monroe and Harkness, Reference Monroe and Harkness2005), even within MZ twin pairs (Kendler and Gardner, Reference Kendler and Gardner2010), and second with findings of no or very low environmental stability after substantial time periods (Kendler et al., Reference Kendler, Neale, Kessler, Heath and Eaves1993; Kendler and Gardner, Reference Kendler and Gardner2010; Torvik et al., Reference Torvik, Rosenstrom, Ystrom, Tambs, Roysamb, Czajkowski, Gillespie, Knudsen, Kendler and Reichborn-Kjennerud2017). Our evidence of stability in MDD is also in agreement with results from a large longitudinal, but not genetically informative Finnish cohort (Rosenstrom et al., Reference Rosenstrom, Jokela, Hintsanen, Pulkki-Raback, Hutri-Kahonen and Keltikangas-Jarvinen2013). The present study is in partial disagreement with a previous study finding 17% stability in environmental risk of MDD over 8 years (Kendler and Gardner, Reference Kendler and Gardner2017), whereas our model implies an environmental stability of only 2% over a similar length of time. The reason for this discrepancy is not apparent, but we note that the present study had a larger sample size, covered a wider age-span, and included both men and women. In any case, these studies and others agree that the stability of risk of MDD over adult life is largely of genetic origin (Burcusa and Iacono, Reference Burcusa and Iacono2007). Our estimate could be interpreted as an average of the durability of life-events, some inducing a risk over shorter and some over longer time spans. We did not detect effects of permanent environmental scarring from severe events, modelled as environmental effects operating throughout the observational period.
Environmental factors shared between twins did not have any significant occasion specific or long-term effects. Behavioral genetic studies have previously found shared environmental influences on depression in childhood, but these become less relevant in adulthood (Bergen et al., Reference Bergen, Gardner and Kendler2007; Lamb et al., Reference Lamb, Middeldorp, van Beijsterveldt, Bartels, van der Aa, Polderman and Boomsma2010). Whereas we cannot rule out that long-term environmental effects exist and are relevant for certain individuals with particularly severe life-events, whether shared or individual-specific, they were not especially important in explaining adult MDD in our sample. As a rule, environmental exposures do not seem to change permanently a person's risk of depression. The findings underline the importance of helping depressed individuals improve their current and future environment. In clinical settings, psychotherapy emphasizing modification of the current environment could be more effective than approaches aimed at understanding past events.
Limitations
The present study has several notable advantages, such as a large, genetically informative, population-based twin sample, with longitudinal clinical data from primary care. Nevertheless, some limitations are noteworthy: First, the sample was based on voluntary participation, and thus subject to nonresponse and possibly associated biases. However, we did not have any attrition after baseline. Second, we only had available data on cases of MDD clinically diagnosed in primary care. Therefore, we could not study sub-clinical levels of depression, individual symptoms, or other conceptualizations of depression. Third, we relied on registry data with diagnostic information based on reimbursement claims from treating physicians in primary care. This implies that in order to be registered, individuals must have sought treatment and received the diagnosis of MDD. Previous research indicates that approximately half of the depressed individuals receive treatment in high-income countries (Thornicroft et al., Reference Thornicroft, Chatterji, Evans-Lacko, Gruber, Sampson, Aguilar-Gaxiola, Al-Hamzawi, Alonso, Andrade, Borges, Bruffaerts, Bunting, de Almeida, Florescu, de Girolamo, Gureje, Haro, He, Hinkov, Karam, Kawakami, Lee, Navarro-Mateu, Piazza, Posada-Villa, de Galvis and Kessler2017). One could, therefore, fear that the health registries are likely to miss many true cases and that the results are not generalizable to depression in general. However, we have previously shown that MDD registered in primary care has a genetic correlation of around 0.80 with both MDD in specialist care and with MDD assessed with structured diagnostic interviews (Torvik et al., Reference Torvik, Ystrom, Gustavson, Rosenstrom, Bramness, Gillespie, Aggen, Kendler and Reichborn-Kjennerud2018). In addition, we found a prevalence similar to major international (Kessler et al., Reference Kessler, Berglund, Demler, Jin, Merikangas and Walters2005; de Graaf et al., Reference de Graaf, ten Have, van Gool and van Dorsselaer2012; Hasin and Grant, Reference Hasin and Grant2015) and previous Norwegian epidemiological studies (Kringlen et al., Reference Kringlen, Torgersen and Cramer2001, Reference Kringlen, Torgersen and Cramer2006), a narrow-sense heritability close to the one reported in a meta-analysis (Sullivan et al., Reference Sullivan, Neale and Kendler2000), and that MDD was associated with expected demographic characteristics (female sex, lower education, lower income, divorce, and single marital status). These observations provide strong indications that the results are representative for individuals with depression. Fourth, it was not feasible to longitudinally model sex differences other than in prevalence, however, univariate analyses on MDD across all time-points suggest no genetic sex differences in our data (Δ−2LL = 2.64, Δdf = 3, p = 0.451).
Conclusion
The genetic and environmental components of clinically assessed MDD exhibit fundamentally different structures. The genetic component is stable over almost 30 years from ages 18 to 45. Therefore, molecular genetic studies may use variable adult age samples to identify genetic risk variants of MDD without introducing genetic heterogeneity in their analyses. The environmental risk factors for MDD were stable over a short span of years with effects rapidly decreasing. We did not detect effects of permanent environmental scarring, as virtually all long-term stability was due to genetic factors. Long-term environmental effects, therefore, do not seem to be important in explaining MDD at the population level.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033291718003550.
Acknowledgements
We wish to thank Kjetil Nordbø Jørgensen, who read and commented on an early version of the manuscript. This project was supported by the Research Council of Norway, grant 240061.
Conflict of interest
None.
Author ORCIDs
Kristin Gustavson 0000-0002-3047-3234, Eivind Ystrom 0000-0003-4390-6171, Nathan Gillespie 0000-0001-8655-2823, Ted Reichborn-Kjennerud 0000-0003-4653-0034, Kenneth S. Kendler 0000-0001-8689-6570