


I. INTRODUCTION
The oxides of cobalt, molybdenum and tungsten, or their mixtures have been widely used as catalysts in reactions of industrial interest such as hydrocarbon oxidation (Matsuura et al., Reference Matsuura, Mizuno and Hashiba1986), dehydrogenation (Mitchell and Wass, Reference Mitchell and Wass2002) and hydrotreating (Zhang et al., Reference Zhang, Duan, Zhao, Wana, Gao, Jiang, Chi and Chuang2010). The last reaction listed here has drawn great attention in the petrochemical field due to its capacity to enhance fuel quality and reduce SOx and NOx emissions (Topsøe et al., Reference Topsøe, Clausen, Massoth, Anderson and Boudart1996).
Currently, mixed oxides can be obtained by several synthesis methods such as co-precipitation (Kolthoff, Reference Kolthoff1932), solid-state (Menon and Delmon, Reference Menon, Delmon, Ertl, Knözinger and Weitkamp1999), sol–gel (Ko, Reference Ko, Ertl, Knözinger and Weitkamp1999), freeze-dried (Vie et al., Reference Vie, Martínez, Sapiña, Folgado and Beltrán2004), and hydrothermal (Rodemerck and Linke, Reference Rodemerck, Linke and De Jong2009). Despite the recognized effectiveness of these synthesis methods, preparation itself of layered materials as precursors of mixed oxides has been also recognized as a promising method (Braterman et al., Reference Braterman, Xu, Yarberry, Auerbach, Carrado and Dutta2004). While the methods presented above usually produce mixed oxides with low-specific areas, their preparation from layered materials as precursors can produce solids with high-specific areas and improved structural properties for catalytic applications.
Lamellar cobalt–molybdates-type ϕ x and ϕ y were synthesized by Pezerat (Reference Pezerat1965). Then, Clearfield et al. (Reference Clearfield, Sims and Gopal1976) and Levin et al. (Reference Levin, Soled and Ying1996) elucidated the structures of phases ϕ x and ϕ y, respectively. Since then, some publications related to the preparation of layered materials, among them layered double hydroxides (LDHs) (Braterman et al., Reference Braterman, Xu, Yarberry, Auerbach, Carrado and Dutta2004), have aroused great importance for their numerous applications in reactions of industrial interest.
This paper reports the synthesis, characterization, and crystallographic parameters of a new trimetallic solid with a formula (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O and isomorphic with the phase ϕ y.
II. EXPERIMENTAL A. SYNTHESIS
A trimetallic layered precursor was synthesized by the hydrothermal method. Three aqueous solutions of (Co(NO3)2•6H2O (Vetec, 98%)), ((NH4)6Mo7O24•4H2O (Merck, 99%)) and ((NH4)10W12O39•5H2O (Alfa Aesar, 99%)) were prepared and then mixed. An NH4OH solution (J.T Baker, 28–30%) was added dropwise until a pH = 8 was attained. A gel with composition Co(NO3)2: 0.1(NH4)6Mo7O24:0.02(NH4)10W12O39:1.4NH4OH:264H2O was obtained and homogenized using magnetic stirring at room temperature for 3 h. Then, it was put in a Teflon reactor at room temperature for 24 h. After crystallization a light purple solid was obtained, recovered by filtration, washed until neutral pH and finally dried at 70 °C.
A. Materials characterization
Powder X-ray diffraction (XRD) data were collected using a Philips X Pert PANalytical Empyrean diffractometer at room temperature. The experimental conditions were: 45 kV, 40 mA, Cu Kα 1/α 2 radiation (λ = 1.5406 Å), 2θ range from 10 to 80°, with 0.02° step size, and 0.5 s per step. Thermogravimetric (TGA) and differential thermal (DTA) analyses were carried out on a TA Instruments equipment, using TGA Hi Res 2950 and DSC 2920, respectively, at a heating rate of 10 °C min−1 under nitrogen atmosphere. An FT–IR spectrum was recorded using an IRAffinity-1-Shimadzu spectrometer. Data were treated with IRSolution software and FT-IR analysis was carried out at room temperature. A tablet-shaped sample was prepared with a 1% potassium bromide composition. Laser Raman spectrum was obtained in a LabRAM HR-UV 800/Jobin-Yvon equipment, with He–Ne (λ = 632 nm) and Ar (λ = 514 nm) lasers. The CCD detector was operated at −70 °C using 10.7 mW power. Metal content analysis for the layered precursor was carried out in Thermo Scientific ICE Series 3000 equipment. The sample was treated with mineral acids (hydrochloric and nitric acids) until complete dissolution was achieved. Interferences associated with each metal were corrected using nitrous oxide and an acetylene flame. Analyses were performed in duplicate.
Crystal system, unit cell parameters and 2θ values were determined using CRYSFIRE (Shirley, Reference Shirley1999) and CHECKCELL (Laugier and Bochu, Reference Laugier and Bochu2000). The data obtained were consistent with the values obtained with EXPO 2009 (Altomare et al., Reference Altomare, Camalli, Cuocci, Giacovazzo, Moliterni and Rizzi2009). Unit cell parameters were refined by CHECKCELL (Laugier and Bochu, Reference Laugier and Bochu2000). Data were also used to determine space groups. Finally, experimental density (Dexp) was estimated by pycnometry in our laboratory. The calculated density (D cal) was determined by the procedure used to measure the properties of crystals as proposed by Stout and Jensen (Reference Stout and Jensen1989).
III. RESULTS AND DISCUSSION
XRD pattern for the trimetallic compound is shown in Figure 1. It observes that the solid is highly crystalline and isostructural with (NH4)HNi2(OH)2(MoO4)2 previously reported by Levin et al. (Reference Levin, Soled and Ying1996) in trigonal symmetry (a = 6.0174 and c = 21.8812 Å).
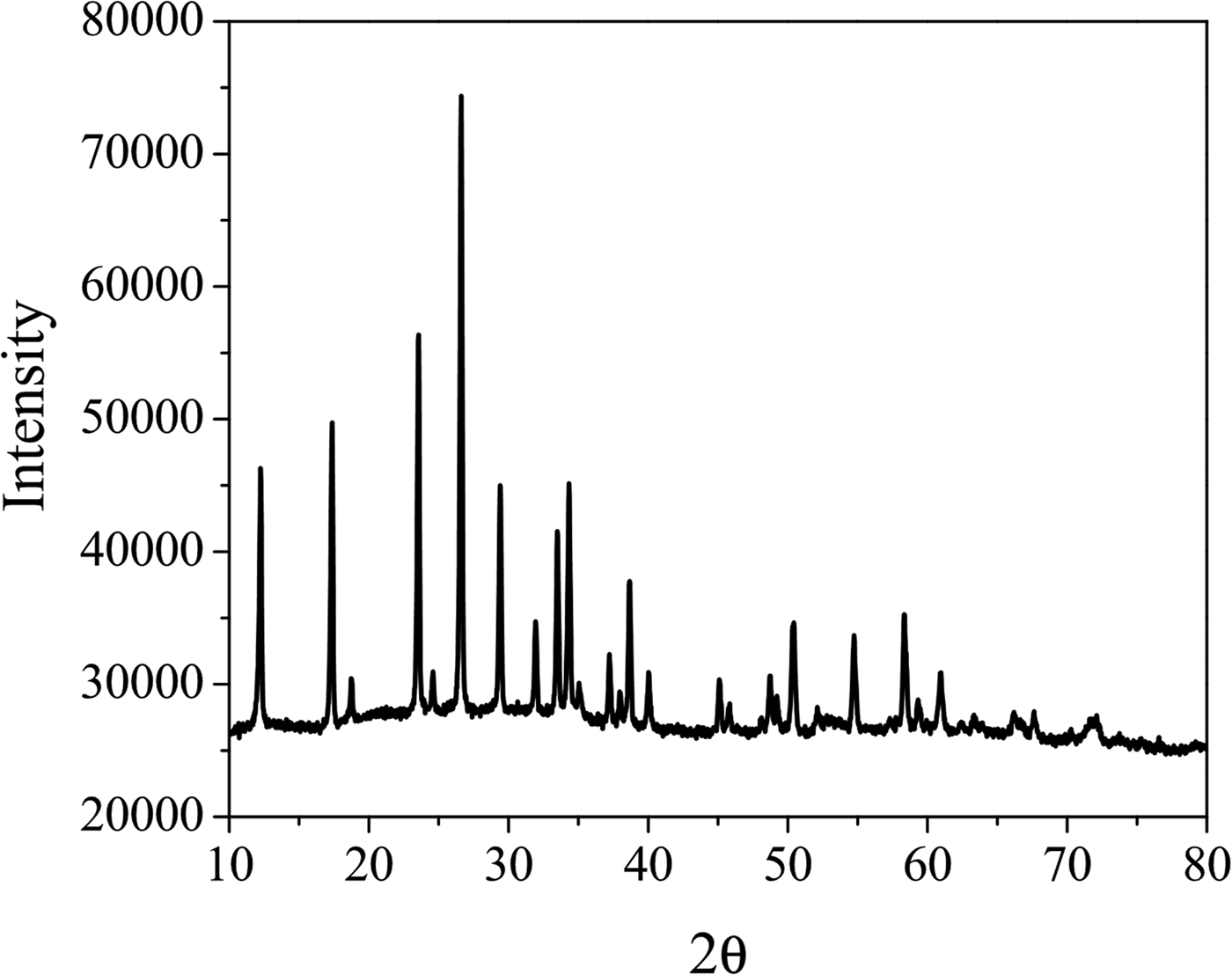
Figure 1. Powder diffraction pattern of (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O.
The chemical composition for the new trimetallic solid was determined using atomic absorption spectroscopy (AA) and thermogravimetric analysis (TGA). The chemical formula for the solid was proposed according to AA and TGA analysis and experimental results are shown in Table I and Figure 2, respectively. In Table I the theoretical composition was calculated from chemical formula proposed, and shows good agreement with experimental results.
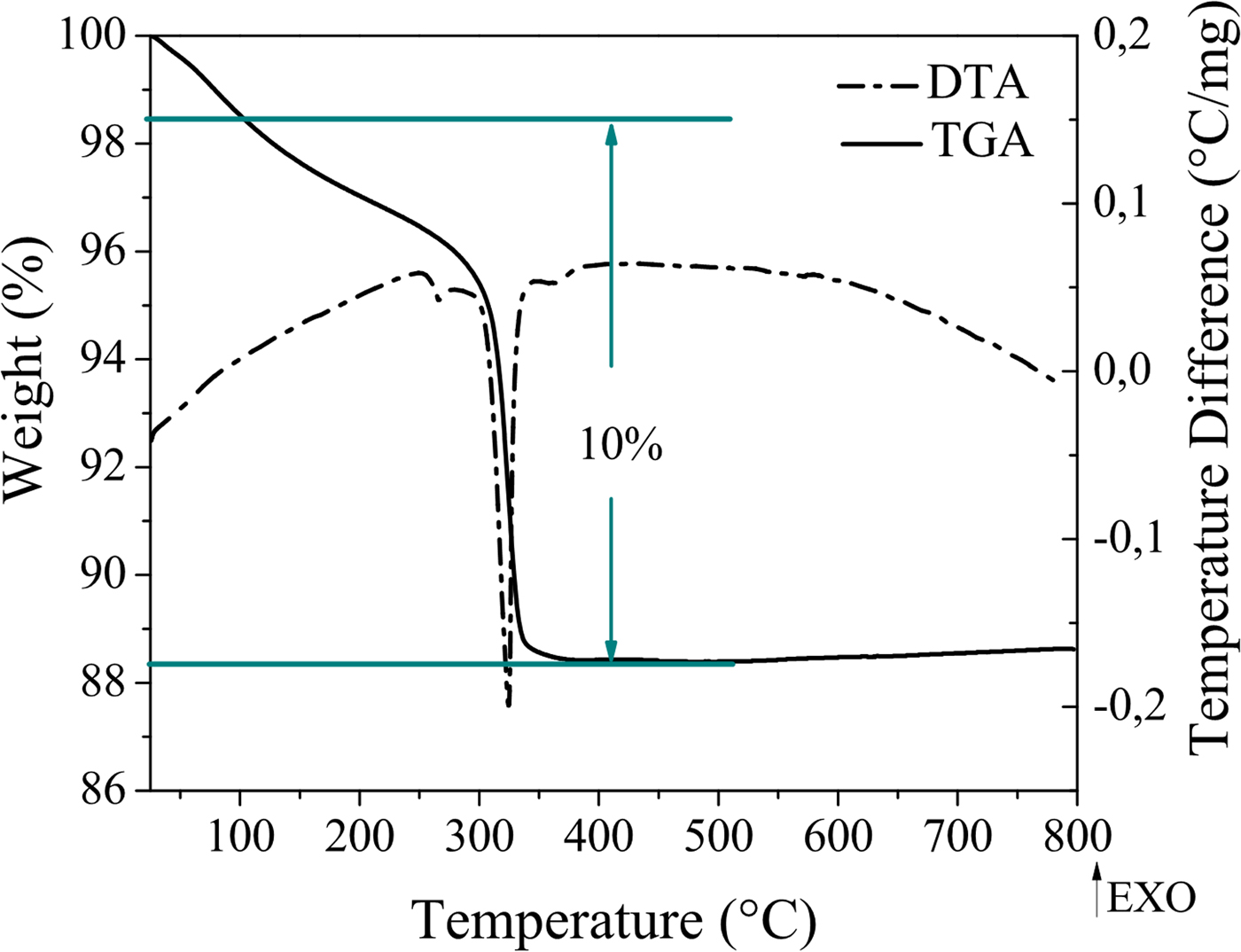
Figure 2. (Color online) TGA and DTA measurements of (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O.
Table I. Elemental compositions for (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O.

Figure 2, TGA and DTA plots showed one endothermic event, which registered a 10% weight loss between 100 and 400 °C for (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O. These events correspond to desorption of water and ammonium shown by the following chemical reaction:
 $$\eqalign{{\rm (N}\hbox{H}_{\rm 4}{\rm )}&\hbox{H}_{\rm 2}\hbox{C}\hbox{o}_{\rm 2}\hbox{O(OH)(Mo}\hbox{O}_{\rm 4}{\rm )}_{{\rm 1}{\rm. 6}}{\rm (W}\hbox{O}_{\rm 4}{\rm )}_{{\rm 0}{\rm. 4}} \cdot \hbox{H}_{\rm 2}\hbox{O} {{\buildrel {\vskip8pt{\hskip-13pt \rm {400}^\circ {\rm C}}} \over { \hscale350%\scale120% \rightarrow}}} \cr & {\rm1}{\rm. 6CoMo}\hbox{O}_{\rm 4}{\rm} + {\rm 0}{\rm. 4CoW}\hbox{O}_{\rm 4} + {\rm N}\hbox{H}_{\rm 3} \uparrow {\rm +3}\hbox{H}_{\rm 2}\hbox{O} \uparrow} $$
$$\eqalign{{\rm (N}\hbox{H}_{\rm 4}{\rm )}&\hbox{H}_{\rm 2}\hbox{C}\hbox{o}_{\rm 2}\hbox{O(OH)(Mo}\hbox{O}_{\rm 4}{\rm )}_{{\rm 1}{\rm. 6}}{\rm (W}\hbox{O}_{\rm 4}{\rm )}_{{\rm 0}{\rm. 4}} \cdot \hbox{H}_{\rm 2}\hbox{O} {{\buildrel {\vskip8pt{\hskip-13pt \rm {400}^\circ {\rm C}}} \over { \hscale350%\scale120% \rightarrow}}} \cr & {\rm1}{\rm. 6CoMo}\hbox{O}_{\rm 4}{\rm} + {\rm 0}{\rm. 4CoW}\hbox{O}_{\rm 4} + {\rm N}\hbox{H}_{\rm 3} \uparrow {\rm +3}\hbox{H}_{\rm 2}\hbox{O} \uparrow} $$The metallic-oxide phases that appear on the right-hand side of the above reaction were identified by the XRD patterns of the solids calcined at 400 °C. Materials treated thermally at 400 °C showed peaks typical of the β phase, as described by Mazzocchia et al. (Reference Mazzocchia, Kaddouri, Anouchinsky, Sautel and Thomas1993). X-ray diffraction pattern of this compound has been included by the International Centre for Diffraction Data (ICDD, Reference Kabekkodu2009) in the Powder Diffraction File (PDF-4) with the number 00-021-0868.
The FT-IR spectrum, as shown in Figure 3, confirmed the presence of ammonium ions in the structure represented by ν 3 (N–H) asymmetric stretching in the 3300–3050 cm−1 region, as well as a characteristic absorption around 1400 cm−1 by bending ν 4 (H–N–H). Absorption bands attributed to the molybdate ion were observed between 925 and 932 cm−1 corresponding to the symmetric stretching vibration mode ν 1 (O–Mo–O) and around 870, 810 and 756 cm−1 to the asymmetric stretching mode ν 3 (Mo–O) (Levin et al., Reference Levin, Soled and Ying1996; Nyquist and Kagel, Reference Nyquist and Kagel1971; Palacio et al., Reference Palacio, Echavarría, Hoyos and Saldarriaga2005). According to these results, the molybdate ion exhibited a tetrahedral environment. Signals attributed to the presence of water were also observed around 1621 and 3300 cm−1 and were associated with H–O–H bending and O–H stretching, respectively (Nyquist and Kagel, Reference Nyquist and Kagel1971; Palacio et al., Reference Palacio, Echavarría, Hoyos and Saldarriaga2005).

Figure 3. FT–IR spectrum of (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O.
As reported by Saleem et al. (Reference Saleem, Aruldhas and Bist1983) and Levin et al. (Reference Levin, Soled and Ying1996), laser Raman analyses have shown that normal modes of the molybdate ion are expected to take place in the regions 750–975 and 250–450 cm−1, corresponding to symmetric stretching and symmetric bending, respectively. The Laser Raman spectrum in this study corroborated this finding (Figure 4) showing two strong signals around 935 and 350 cm−1, which are common for tetrahedral coordination of the molybdate ion, and in accordance with the result obtained by FT-IR.

Figure 4. Laser Raman spectrum of (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O.
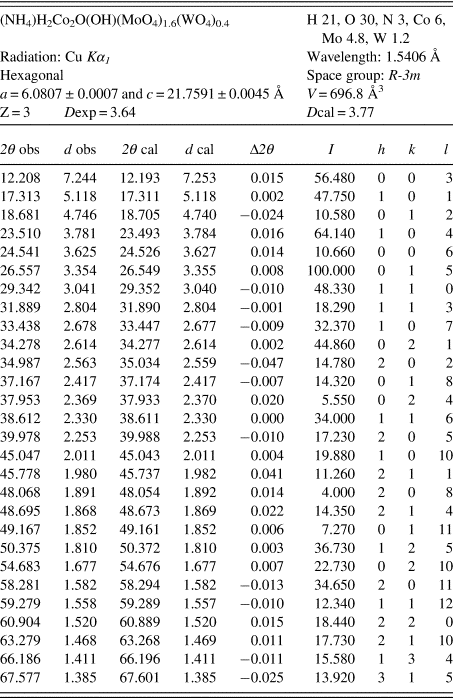
Crystallographic and XRD data of (NH4)H2Co2O(OH)(MoO4)1.6(WO4)0.4•H2O are shown in Table II. The solid crystallized with hexagonal symmetry, space group R-3m with cell parameter values a = 6.0807 and c = 21.7591 Å.
Table II. Diffraction data.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715618000672
ACKNOWLEDGEMENTS
The authors acknowledge financial support from Universidad de Antioquia. S. Amaya acknowledges COLCIENCIAS for her doctoral fellowship.








