Introduction
Embryo cryopreservation is now considered to be a routine procedure for humans and most domestic animals. This refined technique results in high post-thaw survival rates and acceptable implantation and birth rates. Oocyte cryopreservation was developed to improve domestic animal breeding and maintain the biodiversity of endangered animals (Vajta, Reference Vajta2000; Pukazhenthi and Wildt, Reference Pukazhenthi and Wildt2004), and remains a much more challenging procedure. In humans, oocyte cryopreservation enables the reproductive capacity to be retained in cancer patients who may suffer infertility either from disease or treatment (Ali and Sirard, Reference Ali and Sirard2002). It offers women the option to delay reproduction during career development, and avoids the ethical and moral issues associated with embryo cryopreservation. Oocyte freezing also provides the opportunity to create egg banks that are used in some countries to synchronize and optimize donor−recipient timing and standardize the number of eggs being transferred to recipients (Stachecki and Cohen, Reference Stachecki and Cohen2004).
Slow freezing has been widely used and is a well established method to preserve embryos (Veeck et al., Reference Veeck, Amundson, Brothman, DeScisciolo, Maloney, Muasher and Jones1993; Veeck et al., Reference Veeck, Bodine, Clarke, Berrios, Libraro, Moschini, Zaninovic and Rosenwaks2004), but recently vitrification is becoming the method of choice for preserving either cleaved or blastocyst stage embryo due to a higher survival rate (Loutradi et al., Reference Loutradi, Kolibianakis, Venetis, Papanikolaou, Pados, Bontis and Tarlatzis2008). Slow cooling requires low concentrations of cryoprotectants, with controlled stepwise cooling rates of between −0.3 and −2°C/min to prevent formation of intracellular ice crystals (Shaw and Jones, Reference Shaw and Jones2003). Vitrification is a faster, simple procedure and often cheaper than slow cooling and has been reported to reduce chilling injury and osmotic shock, resulting in improved survival rates and embryo development (Lane and Gardner, Reference Lane and Gardner2001; Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005; Gautam et al., Reference Gautam, Verma, Palta, Chauhan and Manik2008; Cobo et al., Reference Cobo, Castello, Vallejo, Albert, de los Santos and Remohi2013a, Reference Cobo, Garcia-Velasco, Domingo, Remohi and Pellicer2013b). In oocyte cryopreservation, more recently better results have been achieved using the vitrification procedure when compared with slow cooling (Lane and Gardner, Reference Lane and Gardner2001; Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005; Gautam et al., Reference Gautam, Verma, Palta, Chauhan and Manik2008; Cao et al., Reference Cao, Xing, Li, Cong, Zhang, Wei and Zhou2009; Cobo et al., Reference Cobo, Garcia-Velasco, Domingo, Remohi and Pellicer2013b; Levi Setti et al., Reference Levi Setti, Porcu, Patrizio, Vigiliano, de Luca, d’Aloja, Spoletini and Scaravelli2014). However, when compared with slow cooling, vitrification often requires twice the concentration of permeating cryoprotectants in the cryo-medium and can be lethal for oocytes if the exposure time is too long, the volume is too large or the rates of cooling are too slow. Ideally, exposure of oocytes to the vitrifying solution should be less than a minute to minimize the toxic effects and the cooling rate should be around −50,000 to −200,000°C/min to prevent the formation of intra- and extra-cellular ice crystals (Shaw and Jones, Reference Shaw and Jones2003; Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005).
The permeating capacity and toxicity of intracellular cryoprotectants vary among the different agents. For example, 1,2 propanediol and 2,3 butanediol were less cytotoxic than dimethyl sulfoxide (DMSO) and ethanediol when umbilical vein endothelial cells were exposed to these agents (Wusteman et al., Reference Wusteman, Pegg, Robinson, Wang and Fitch2002). Despite this, DMSO achieves a better solid glass state upon vitrification when compared with two other commonly used cryoprotectants, acetamide or ethylene glycol (EG) (Fahy et al., Reference Fahy, Wowk, Wu and Paynter2004). To date, EG or a combination of DMSO and EG or PROH and EG are the most common cryoprotectants either commercially or in house production to be used in mouse, cattle, goat, lamb, fox, cat and human oocyte and embryo cryopreservation (Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005; Succu et al., Reference Succu, Leoni, Bebbere, Berlinguer, Mossa, Bogliolo, Madeddu, Ledda and Naitana2007a; Gautam et al., Reference Gautam, Verma, Palta, Chauhan and Manik2008; Berthelot-Ricou et al., Reference Berthelot-Ricou, Perrin, di Giorgio, de Meo, Botta and Courbiere2013; Larman and Gardner, Reference Larman and Gardner2014; Fernandez-Gonzalez and Jewgenow, Reference Fernandez-Gonzalez and Jewgenow2016; Cao et al., Reference Cao, Li, Xue, Wang, Zhaow, Du, Yang and Yue2017).
Embryos derived from vitrified oocytes differ in their development between species. High survival, and high blastocyst and pregnancy rates have been reported in the mouse (Lane and Gardner, Reference Lane and Gardner2001), but in other animals such as lamb (Succu et al., Reference Succu, Leoni, Bebbere, Berlinguer, Mossa, Bogliolo, Madeddu, Ledda and Naitana2007a, Reference Succu, Leoni, Berlinguer, Madeddu, Bebbere, Mossa, Bogliolo, Ledda and Naitana2007b, Reference Succu, Bebbere, Bogliolo, Ariu, Fois, Leoni, Berlinguer, Naitana and Ledda2008), buffalo (Gautam et al., Reference Gautam, Verma, Palta, Chauhan and Manik2008; Liang et al., Reference Liang, Srirattana, Phermthai, Somfai, Nagai and Parnpai2012), pig (Somfai et al., Reference Somfai, Ozawa, Noguchi, Kaneko, Kuriani Karja, Farhudin, Dinnyes, Nagai and Kikuchi2007), goat (Begin et al., Reference Begin, Bhatia, Baldassarre, Dinnyes and Keefer2003), fox (Cao et al., Reference Cao, Li, Xue, Wang, Zhaow, Du, Yang and Yue2017), and cat (Fernandez-Gonzalez and Jewgenow, Reference Fernandez-Gonzalez and Jewgenow2016) embryo development rates still remain low, probably due to a variety of factors including disruption of meiotic spindles and chromosomes (Albarracin et al., Reference Albarracin, Morato, Izquierdo and Mogas2005; Succu et al., Reference Succu, Leoni, Bebbere, Berlinguer, Mossa, Bogliolo, Madeddu, Ledda and Naitana2007a, Reference Succu, Leoni, Berlinguer, Madeddu, Bebbere, Mossa, Bogliolo, Ledda and Naitana2007b; Berthelot-Ricou et al., Reference Berthelot-Ricou, Perrin, di Giorgio, de Meo, Botta and Courbiere2013; Cao et al., Reference Cao, Li, Xue, Wang, Zhaow, Du, Yang and Yue2017), parthenogenetic activation (Tian et al., Reference Tian, Yan, Yang, Zhou, Yang and Zhu2007), premature cortical granule rupture and zona hardening, which decreased sperm penetration at fertilization (Fuku et al., Reference Fuku, Xia and Downey1995; Tian et al., Reference Tian, Yan, Yang, Zhou, Yang and Zhu2007), and decreased glutathione production (Cao et al., Reference Cao, Li, Xue, Wang, Zhaow, Du, Yang and Yue2017). The common method for insemination of vitrification-warmed human oocytes is intracytoplasmic sperm injection (ICSI), which avoids zona hardening issues (Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005; Chang et al., Reference Chang, Shapiro, Bernal, Wright, Kort and Nagy2008; Cao et al., Reference Cao, Xing, Li, Cong, Zhang, Wei and Zhou2009; Cobo et al., Reference Cobo, Castello, Vallejo, Albert, de los Santos and Remohi2013a). However, in animal studies, in which large numbers of oocytes are often processed, in vitro fertilization (IVF) remains a popular method for insemination of vitrification-warmed oocytes (Succu et al., Reference Succu, Leoni, Berlinguer, Madeddu, Bebbere, Mossa, Bogliolo, Ledda and Naitana2007b, Reference Succu, Bebbere, Bogliolo, Ariu, Fois, Leoni, Berlinguer, Naitana and Ledda2008; Gautam et al., Reference Gautam, Verma, Palta, Chauhan and Manik2008). A few large studies in cattle and humans have reported high fertilization rates, blastocyst rates and pregnancies from post-vitrified oocytes by either IVF or ICSI (Vajta et al., Reference Vajta, Holm, Kuwayama, Booth, Jacobsen, Greve and Callesen1998; Papis et al., Reference Papis, Shimizu and Izaike2000; Tong et al., Reference Tong, Wu, Jin, Luo, Luan and Liu2012; Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005) but most results have been variable and no universal method has yet been established.
The objectives of this study were to evaluate the survival rate of mouse and lamb oocytes after exposure to different combinations of commonly used cryoprotectants and vitrification itself, fertilizing ability, developmental competence of fertilized or activated lamb and mouse vitrified-warmed oocytes and the incidence of cortical granule exocytosis and zona hardening in lamb oocytes.
Materials and methods
Chemicals
All chemicals and reagents were purchased from Sigma Chemical Co. (St. Louis, MO, USA) unless otherwise stated.
Oocyte collection, selection and in vitro maturation (IVM): lamb
As our laboratory did not have the facilities to perform in vivo maturation in lamb, those oocytes were in vitro matured. Ovaries were obtained from lambs (3–9 months) within 2 h of slaughter. Ovaries were washed three times in pre-warmed saline supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin (GIBCO, New Zealand). Oocytes were aspirated from follicles of diameter 2–6 mm into tissue culture medium (TCM)-199 handling medium (MP Biomedicals, Solon, OH, USA), supplemented with 2.5 mM sodium hydrogen carbonate, 1 mM glutamine, 2 mM sodium pyruvate, 22.5 mM HEPES, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2% (v/v) lamb serum (GIBCO) and supplemented with 5000 IU/ml heparin (Pfizer, WA, USA). Oocytes with at least two layers of cumulus cells and homogenous cytoplasm were washed in TCM handling medium and cultured under mineral oil in 500 μl IVM medium (M-199 with 25 mM Na bicarbonate, 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml follicle-stimulating hormone (FSH) (Folltropin V, Bioniche, Canada), 5 μg/ml luteinising hormone (LH) and 20% (v/v) lamb serum) in four-well Petri dishes (Nunclon, Nalge Nunc International, Denmark) for 24–26 h in a Mini-incubator (MINC, COOK Medical, Brisbane, Australia) using a humidified atmosphere of 6% CO2, 5% O2 in air at 39°C.
Mature oocyte collection: Mouse
F1 C57B/J × CBA mice aged 4–5 weeks were superovulated by intraperitoneal (IP) administration of 7.5 IU of pregnant mare serum gonadotrophin (PMSG; Folligon; Intervet, Australia) followed 48–60 h later with intraperitoneal (i.p.) 7.5 IU human chorionic gonadotrophin (hCG, Chorulon, Intervet). Mice were killed by cervical dislocation 13–14 h post hCG, and mature cumulus–oocyte complexes were collected from the oviduct into HEPES–KSOM (kalium simplex optimized medium) supplemented with 0.5% (v/v) non-essential amino acid (GIBCO), 1% (v/v) essential amino acid (GIBCO) and 0.3% (w/v) BSA (GIBCO), modified from Summers et al. (Reference Summers, Bhatnagar, Lawitts and Biggers1995). Cumulus cells were denuded by treatment with 60 IU hyaluronidase in HEPES–KSOM for 30–60 s before transfer into modified Tyrode’s (mT6) medium, adapted from Fraser (Reference Fraser1984).
Oocyte vitrification and warming
Vitrification and warming solutions were prepared in HEPES–KSOM medium for mouse and HEPES–synthetic oviductal fluid (SOF) for lambs. Three vitrification solutions (VS) were tested: 2.3 M dimethylsulfoxide (DMSO) + 3 M EG; 6 M EG only; 2.3 M 1,2 propanediol (PROH) + 3 M EG. All VS were supplemented with 0.75 M sucrose. Vitrification was performed at 37°C. Corresponding equilibration solutions (ES) contained half the stated concentration of cryoprotectants and lacked sucrose. Denuded oocytes were placed in ES, equilibrated for 3 min, transferred to VS, then with minimal solution (less than 3 μl) placed on a fibreplug, touched to a pre-cooled steel block in liquid nitrogen and sealed in a pre-cooled straw (CVM kit, Cryologic, Victoria, Australia) for storage in liquid nitrogen. The time taken from leaving ES to vitrification on the block was between 30 to 40 s. Vitrified oocytes were warmed at 37°C serially through three solutions (0.3 M, 0.25 M, 0.15 M sucrose in HEPES–SOF medium for lamb and HEPES–KSOM for mouse) for 5 min each, then washed in fresh HEPES handling medium. All VS (PROH and EG) were from the same batch, except for DMSO in which a fresh ampoule was opened every 3 weeks. Control oocytes were left fresh without exposure to cryoprotectants and vitrified. Exposed groups of cryoprotectants consisted of oocytes that were exposed to the cryoprotectants, but not vitrified to observe the toxicity of cryoprotectants on oocyte survival only. Vitrified-warmed oocytes were left for 2 h before fertilization.
In vitro fertilization (IVF) and in vitro culture (IVC)
Lambs
In total, 20–30 matured oocytes were transferred into 500 μl modified SOF, adapted from Tervit et al. (Reference Tervit, Whittingham and Rowson1972), supplemented with 2% (v/v) lamb serum. Frozen ram sperm pellets were thawed in a 5-ml glass tube whilst shaking at 37°C water bath for 10 s then layered on a discontinuous 45%/90% Percoll gradient in 12-ml Falcon tubes (Becton Dickinson, Franklin Lakes, NJ, USA) and centrifuged at 600 g for 15 min. The supernatant was removed and the pellet was diluted with HEPES–SOF medium and centrifuged at 300 g for 6 min. Oocytes were inseminated with 2 × 106 motile spermatozoa/ml for 18 h in an incubator with an humidified atmosphere of 6% CO2, 5% O2 in air at 39°C. Oocytes were cleaned free of cumulus and attached spermatozoa were removed by manual pipetting. Some presumptive zygotes were fixed and stained to observe normal, abnormal and parthenogenetic activation and the rest of oocytes were transferred into Sydney IVF cleavage medium (COOK® Medical, cat. no. K-SICM 50, Brisbane, Australia) to observe oocyte developmental competence. On day 3, cleaved oocytes with four or more blastomeres were transferred into Sydney IVF blastocyst medium (COOK® Medical, cat. no. K-SIBM 50). Cleavage and blastocyst rates were assessed at day 2 and day 8 after insemination.
Mouse
Fresh spermatozoa were collected from the cauda epididymis of C57B/J × CBA males, placed into 1 ml mT6 in 12 ml Falcon tubes and incubated at 37°C, in 5% CO2 in air for 30–60 min. Oocytes were inseminated with 2.5 × 105 motile spermatozoa/ml in 50 μl mT6 droplets in 35 mm Falcon dishes, covered with mineral oil for 4 h in an incubator under an atmosphere of 5% CO2 in air, at 37°C, and then transferred into Sydney IVF cleavage medium for 2 days and into Sydney IVF blastocyst medium on day 3. Cleavage and blastocyst rates were assessed at day 1 and day 4 after insemination. Blastocysts derived from control and vitrified oocytes were assessed for cell count on day 4. Those blastocysts were incubated with Hoechst 33342 stain (5 μg/ml in HEPES–KSOM) for 30 min at 37°C and assessed immediately under UV fluorescence.
Staining for lamb pronuclei (for evidence of normal, abnormal and parthenogenetic activation)
At 18 h post-fertilization, lamb presumptive zygotes were stained with Hoechst 33342 dye, 5 μg/ml in HEPES–SOF medium for 30 min at 39°C, then assessed by UV fluorescence microscopy (Leica DMR Upright Fluorescence Microscope, Germany). Oocytes were identified as normally and abnormally fertilized and parthenogenetic activation as previously described (Han and Gao, Reference Han and Gao2013; Liang et al., Reference Liang, Srirattana, Phermthai, Somfai, Nagai and Parnpai2012; Tian et al., Reference Tian, Yan, Yang, Zhou, Yang and Zhu2007; Gomez et al., Reference Gomez, Catt, Gillan, Evans and Maxwell1997). Normal fertilization was identified by the appearance of two polar bodies (2 PB) and two pronuclei (2 PN). The oocytes were scored as parthenogenetic when there was no sperm observed but a single pronucleus or 2 PN in the cytoplasm with only 1 PB or no PB. Oocytes with penetrated sperm or a decondensed sperm head in the cytoplasm or with 2 PB and more than 2 PN were assessed as having abnormal fertilization. Oocytes with a metaphase spindle and 1 PB but no evidence of sperm penetration were classified as unfertilized oocytes (Fig. 1C). Germinal vesicle and metaphase I stage oocytes were identified and excluded.
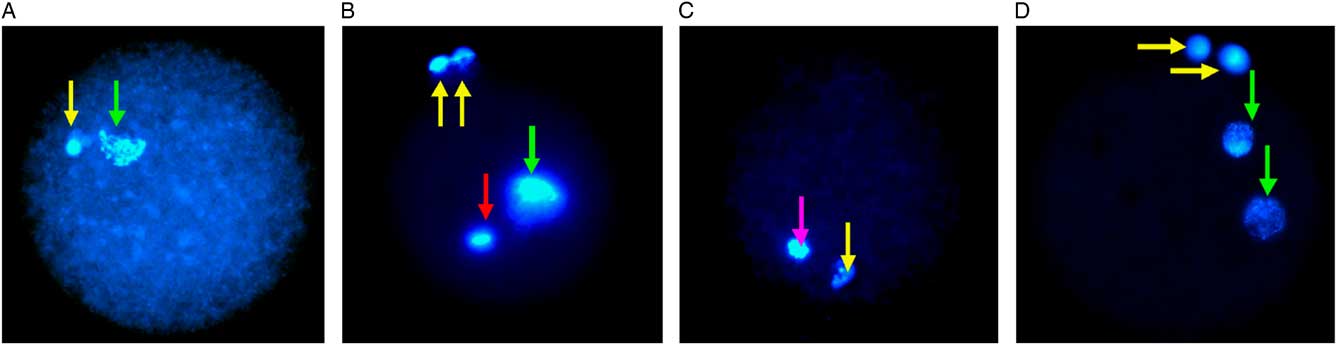
Figure 1 Fluorescence microscopy of vitrified-warmed lamb oocytes after insemination. The positions of pronuclei (PN) are marked with a green arrow and the polar body (PB) with a yellow arrow. (A) Parthenogenetic activation demonstrated by the presence of 1 PN and 1 PB. (B) Abnormal fertilization, with 1 PN, 2 PB and a sperm head is indicated by a red arrow. (C) Mature MII unfertilized oocyte with 1 PB and meiotic spindle is indicated by a pink arrow. (D) Normal fertilization indicated by 2 PN and 2 PB.
Parthogenetic activation on mouse oocytes
To observe parthenogenetic activation in the mouse, oocytes were cultured for 1 day without insemination and recorded as parthenogenetic embryos if cleavage occurred within 24 h. As a positive control, mouse oocytes were activated with parthenogenetic medium (10 mM SrCl2, 5 μg/ml cytochalasin B and 10 mM EGTA in HEPES–KSOM) as described by Kishigami and Wakayama (Reference Kishigami and Wakayama2007). Briefly, denuded mature oocytes were transferred into parthenogenetic medium and cultured for 6 h then washed in HEPES–KSOM medium before being transferred to Sydney IVF cleavage and blastocyst medium.
Staining for cortical granules
In vitro matured lamb oocytes, either unfrozen (control group) or 1–4 h post-warming, were removed of their zona pellucida using 0.5% (w/v) pronase in HEPES–SOF medium at 39°C. Oocytes were continuously observed for zona dissolution using a stereomicroscopy and the time taken for complete dissolution was recorded. The zona-free oocytes were washed in 0.01% (v/v) Triton-X in HEPES–SOF medium then fixed in 4% (w/v) paraformaldehyde. After washing three times (5 min each) in blocking solution containing 10 mM glycine in HEPES–SOF medium, oocytes were permeabilized with 0.1% (v/v) Triton-X 100, then incubated for 30 min at 39°C in 1 μmol/l fluorescein isothiocyanate conjugated to Lens culinaris agglutinin (FITC–LCA) and washed four times for 5 min each in blocking solution and were mounted in glycine solution in twin cavity slides for observation using confocal laser scanning microscopy (CLSM, Leica TCS NT upright equipped by CLSM, Germany). Next, 5 μm sections were imaged from the surface to a depth of 20 μm. The grey level intensity of FITC–LCA was measured at 20 μm using Image J (National Institutes of Health, USA) and calculated relative to surface area.
Statistical analysis
Data for survival, normal and abnormal fertilization, parthenogenetic activation and embryo development were analysed using Fisher’s exact test. Blastocyst total cell number and zona pellucida dissolving time were assessed by paired t-test. A non-parametric repeated measures analysis of variance (ANOVA) was used to compare measured grey level intensities for FITC–LCA staining using Statgraphics software (Statpoint Technologies Inc., Virginia, USA). Experiments were replicated at least three times in each treatment group. All graphed data were presented as mean ± standard error of the mean (SEM).
Results
Effect of different cryoprotectants and vitrification on mouse and lamb oocyte survival rates
Survival rates of lamb oocytes after passing through the warming solutions for all exposed groups were >90%. Post-warming survival rates (vitrified groups) for lamb oocytes vitrified in DMSO + EG (79.5%), EG (67.9%), and PROH + EG (77.6%) were significantly lower (P <0.05) than for exposed oocytes (97.1, 90 and 97.2%, respectively). DMSO + EG and PROH + EG resulted in better post-warm survival rates than EG (79.5 and 77.6% compared with 67.9% respectively, P <0.05; Table 1).
Table 1 Effect of different cryoprotectants and vitrification on survival and degeneration of lamb oocytes

* From the total number of inseminated oocytes from vitrified groups, some were cultured to observe embryo development and the rest were fixed and stained to assess normal and abnormal fertilization status and parthogenetic activation. Control-112 cultured, 78 fixed and stained; DMSO + EG-85 cultured, 51 fixed and stained; EG-61 cultured, 45 fixed and stained; PROH + EG-85 cultured, 57 fixed and stained.
a,b,c Different superscripts within columns denotes statistical difference (P <0.05).
N/A: not applicable.
In contrast with lamb oocytes, mouse oocytes vitrified in PROH + EG had a lower survival rate than those in DMSO + EG (32.7% vs 87.8%, P <0.01) and 10 times greater degeneration rate following culture (15.2% vs 1.6%, P <0.01). A similar result was obtained for oocytes exposed to PROH + EG and DMSO + EG (survival 25.8% vs 93%, P <0.01; degeneration rate following culture 37.5% vs 2.5%, P <0.01, respectively; Table 2). Vitrification using EG alone was only tested in lambs and was not tested for embryo development in the mouse, due to poor survival rates (5/34, 14.7%) in preliminary experiments.
Table 2 Effect of different cryoprotectants and vitrification on survival and degeneration of mouse oocytes

* From 46 fresh control oocytes, 36 were fertilized to observe embryo development and 10 oocytes were used as negative controls to observe parthenogenetic activation in mouse oocytes. In the DMSO + EG vitrified group, 87 oocytes were inseminated and cultured and 35 oocytes were not inseminated to assess parthenogenetic activation on day 1.
a,b Different superscripts within columns denote statistical difference (P <0.01).
Effect of different cryoprotectants and vitrification on normal and abnormal fertilization, and parthenogenetic activation in mouse and lamb oocytes
In lambs, normal fertilization rates in DMSO + EG (17.6%), EG (22.2%), PROH + EG (10.5%) were significantly lower than in the controls (47.4%, P <0.01; Table 3; Fig. 1D). The incidence of parthenogenetic activation was significantly higher in DMSO + EG (35.3%, P <0.01), 24.4% in EG (P <0.05), 26.3% in PROH + EG (P <0.01) compared with controls (9%; Table 3; Fig. 1A). There was no significant difference in abnormal fertilization rate including polyspermia in treatment groups compared with controls (Table 3; Fig. 1B). In contrast with lambs, only one of 35 mouse oocytes (2.9%) in DMSO + EG group developed into two cells without insemination, but did not develop to the blastocyst stage; In contrast, in the positive control of parthenogenetic activation, 100% (10/10) of oocytes grew into two cells and 70% (7/10) developed to blastocysts (Table 4).
Table 3 Effect of different cryoprotectants and vitrification on normal and abnormal fertilization, and parthenogenetic activation of lamb oocytes

Different superscripts within columns denote statistical difference: a vs b: P <0.01, c vs d: P <0.05.
Table 4 Effect of different cryoprotectants, vitrification and induced chemical activation in the absence of sperm on mouse oocyte development

Effect of different cryoprotectants and vitrification on lamb and mouse oocyte developmental competence
Post IVF, cleavage rate of all vitrified-warmed lamb oocytes was similar and statistically lower than control oocytes (DMSO + EG 32.9%, EG 32.8% and PROH + EG 21.2% vs control 84.8%, P <0.01), and blastocyst development from cleaved embryos was lower for vitrified groups (PROH + EG and EG) <6% compared with the control 38.9% (P <0.01; Table 5). In contrast with lambs, cleavage rate of vitrified-warmed oocytes with PROH + EG had a 30% lower cleavage compared with DMSO + EG (P <0.01, Table 6). A trend for reduced blastocyst development in the PROH + EG group was also evident, however this was not significant as only small sample numbers could be analyzed due to extensive degeneration and poor cleavage rates for these oocytes. Compared with control oocytes, the proportions of embryos from PROH + EG and DMSO + EG groups that cleaved and formed blastocysts was significantly lower (P <0.01 and P <0.05 respectively); with significantly fewer cell number of blastocysts in the DMSO + EG treatment group (33 ± 3.1) compared with the control group (42 ± 1.5, P <0.05; Table 6).
Table 5 Effect of different cryoprotectants and vitrification on lamb oocyte developmental competence

Different superscripts within columns denote statistical difference (P <0.01).
Table 6 Effect of different cryoprotectants and vitrification on mouse oocyte developmental competence

Different superscripts within columns denote statistical difference: a vs b, c: P <0.01, b vs c: P <0.01, d vs e: P <0.05, f vs g: P <0.05.
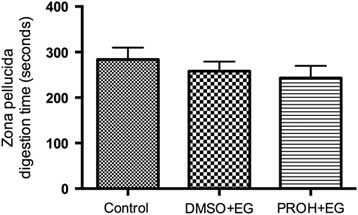
Cortical granule exocytosis and zona dissolution times in post-warmed vitrified lamb oocytes
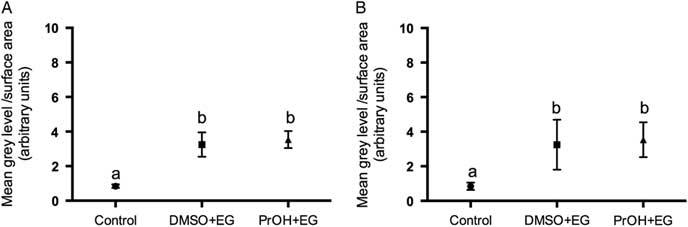
Two treatment groups; DMSO + EG and PROH + EG were observed for cortical granules exocytosis. No differences were found in the timing of the zona pellucida dissolution between fresh and vitrified lamb oocytes (Fig. 4). The relative grey level intensity for FITC–LCA in oocyte cross-sections, each measured at a depth of 20 μm, was around three times greater in vitrified lamb oocytes compared with the controls (Fig. 2; P <0.05). Overall, the higher fluorescent intensity in vitrified oocytes represented diffuse staining throughout the ooplasm (Fig. 3). Cytoplasmic staining was not apparent in control oocytes, which demonstrated only cortical fluorescence, around 2 μm beneath the oolemma (Fig. 3A).
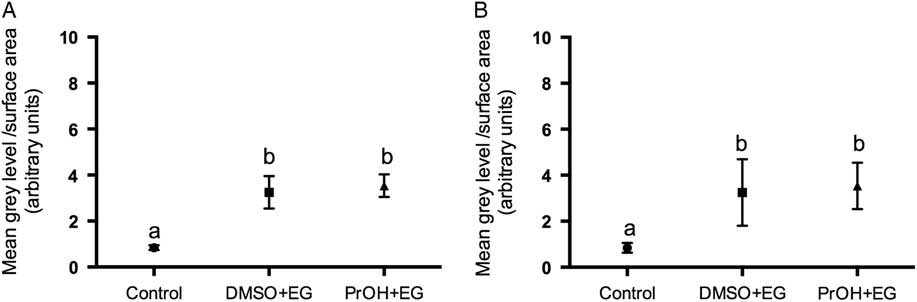
Figure 2 Grey level intensity of fresh control and vitrified-warmed oocytes (DMSO + EG and PROH + EG) stained with fluorescein isothiocyanate conjugated to Lens culinaris agglutinin (FITC–LCA) to demonstrate the presence of α-d-mannose and α-d-glucose in cortical granules. Different superscripts denote significant differences (P <0.05).
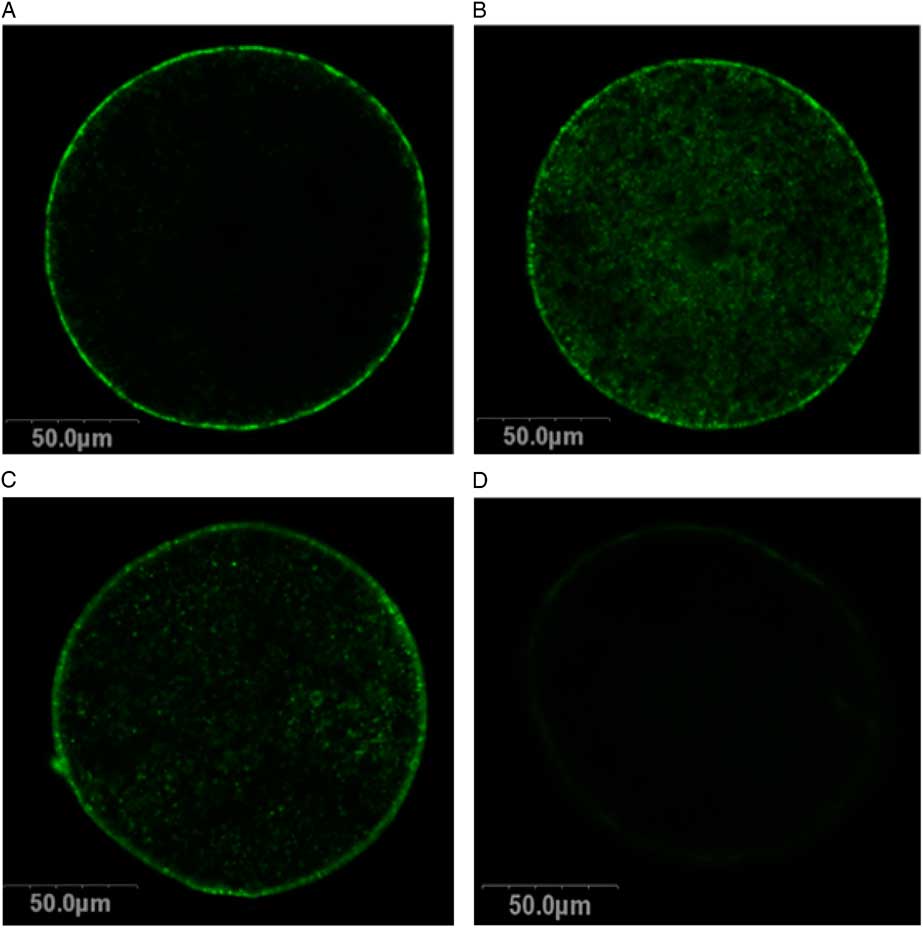
Figure 3 Cortical granules in lamb oocytes. Cross-sections of lamb oocytes labelled with FITC–LCA to stain cortical granules and imaged at a depth of 20 μm from the plasma membrane. Cortical granules in control oocytes (A) were identified at the cortical edge of control oocytes, about 2 μm beneath the oolemma with very little staining within the cytoplasm. Oocytes vitrified using DMSO + EG (B) and PROH + EG (C) exhibit diffuse cortical granule staining throughout the cytoplasm. Negative controls (D) exhibited no autofluorescence.

Figure 4 Zona pellucida digestion time (s) for lamb oocytes. Zona pellucida dissolving time (mean ± standard error of the mean (SEM)) in fresh control and vitrified-warmed lamb oocytes (DMSO + EG and PROH + EG) are not significantly different between groups.
Discussion
Mouse oocytes were extremely compromised in VS containing EG alone, and due to very poor survival rates in preliminary experiments, vitrification in EG was not tested further. Mouse oocyte survival using DMSO + EG during vitrification was significantly higher than those vitrified with PROH + EG. Combined with the observation that similar survival rates were obtained using the same cryoprotectant treatments without vitrification, this suggests that PROH, not the vitrification process itself, was detrimental in the mouse model. In general, successful vitrification requires permeating cryoprotectant concentrations to be around 4.5 M (Shaw and Jones, Reference Shaw and Jones2003). Using 2.3 M PROH satisfied this requirement when combined with 3 M EG. Nevertheless, 2.5 M PROH has been reported to cause degeneration of >80% of mouse oocytes (Mullen, Reference Mullen2007), and only 32% oocytes survived after exposure to 1.5 M PROH with 38% cleavage rate (Gook et al., Reference Gook, Osborn and Johnston1993). Of oocytes that were subsequently slow cooled, only 4% survived and none cleaved following insemination. However, Huang et al. (Reference Huang, Chen, Park, Tan and Chian2008) reported that 91.8% of oocytes survived after vitrified with 2 M PrOH + 2.6 M EG, but only 30.5% developed into blastocysts (Huang et al., Reference Huang, Chen, Park, Tan and Chian2008). The mechanism behind toxic injury from PROH in the mouse is not fully understood. Propanediol is known to decrease the viability of oocytes by altering the oocyte proteome due to a prolonged elevation of intracellular calcium levels in mouse oocytes (Larman et al., Reference Larman, Katz-Jaffe, Sheehan and Gardner2007). The cytoskeletal actin molecule in mouse oocytes was also found to be sensitive to PROH, causing an alteration of the actin molecule and inducing fragility (Maro et al., Reference Maro, Johnson, Pickering and Flach1984). Moreover, it has been shown that VS containing PROH induced more DNA damage on mouse oocytes compared with DMSO (Berthelot-Ricou et al., Reference Berthelot-Ricou, Perrin, di Giorgio, de Meo, Botta and Courbiere2013). As vitrified mouse oocyte in the DMSO + EG group has higher embryo development compared with the PROH + EG group, it was demonstrated that the former combination is more suitable for preserving mouse oocytes.
Unlike mice, oocytes collected from abattoir-sourced lamb ovaries did not demonstrate a specific toxicity to DMSO, PROH or EG. Survival rates after exposure to VS without vitrification were similar using either PROH + EG or DMSO + EG, and both demonstrated better post-warm survival than EG alone. It has been proposed that a combination of cryoprotectants was more successful than using a single agent (Van der Elst et al., Reference Van der Elst, Van den Abbeel, Nerinckx and Van Steirteghem1992), therefore reducing the required concentration for individual cryoprotectants, and possibly reducing overall toxicity. Nevertheless the rate of lamb oocyte lysis following the vitrification process was high for all groups. Lamb oocytes are remarkably different from murine oocytes, they are larger, have a higher intracellular lipid content and have poor post-warming recovery of meiotic spindles (Succu et al., Reference Succu, Leoni, Bebbere, Berlinguer, Mossa, Bogliolo, Madeddu, Ledda and Naitana2007a, Reference Succu, Leoni, Berlinguer, Madeddu, Bebbere, Mossa, Bogliolo, Ledda and Naitana2007b). The sensitivity of lamb oocytes to cooling injury may result from destruction of intracellular lipids through a mechanism of lipid phase transition (LPT) in which membranes, as they cool and reheat, will change their form. For example, unsaturated fatty acids in non-lamellar forming lipids assume a hexagonal II phase structure that alters membrane function and leads to ion leakage and cell death (Arav and Zvi, Reference Arav and Zvi2008; Quinn, Reference Quinn1985).
Parthenogenetic activation was investigated in mouse oocytes vitrified using DMSO + EG and no evidence for a greater incidence of parthenogenetic activation was found. In contrast, higher parthenogenetic activation rates have been reported in mouse oocytes vitrified using PROH compared with DMSO as a cryoprotectant (Shaw and Trounson, Reference Shaw and Trounson1989). Chemicals containing hydroxyl (–OH) groups such as PROH are reported to stimulate parthenogenetic activation of mouse oocytes (Shaw and Trounson, Reference Shaw and Trounson1989; Van der Elst et al., Reference Van der Elst, Van den Abbeel, Nerinckx and Van Steirteghem1992). In this experiment, the low numbers of oocytes surviving exposure to, or vitrification with, PROH precluded examination of parthenogenetic activation. By contrast, lamb parthenogenetic activation rate was significantly increased in all treatment groups. Moreover, there was an increased trend of parthenogenetic activation for DMSO + EG than for EG alone. This is supported by previous reports of a higher incidence of parthenogenetic activation in lamb oocytes using DMSO + EG (63.9%) compared with untreated oocytes (8.2%) (Tian et al., Reference Tian, Yan, Yang, Zhou, Yang and Zhu2007).
In both species, the number of embryos that reached the two-cell stage and developed into blastocysts after vitrification and insemination was significantly higher for unfrozen oocytes than for all vitrified groups. The only available source of ovine oocytes was from pre-pubertal lamb ovaries, therefore this may have been the reason for the relatively low blastocyst development. It has been demonstrated that there was lower blastocyst development in younger lambs compared with adult ewes (Succu et al., Reference Succu, Leoni, Berlinguer, Madeddu, Bebbere, Mossa, Bogliolo, Ledda and Naitana2007b). The blastocyst rate post-vitrified-warmed adult sheep oocytes was reported to be around 0−17% (Marco-Jimenez et al., Reference Marco-Jimenez, Berlinguer and Leoni2012; Succu et al., Reference Succu, Leoni, Bebbere, Berlinguer, Mossa, Bogliolo, Madeddu, Ledda and Naitana2007a, Reference Succu, Bebbere, Bogliolo, Ariu, Fois, Leoni, Berlinguer, Naitana and Ledda2008) and 0% from young lamb oocytes (Succu et al., Reference Succu, Leoni, Berlinguer, Madeddu, Bebbere, Mossa, Bogliolo, Ledda and Naitana2007b). From this present study, normal fertilization rate of vitrified-warmed oocytes was low compared with fresh controls. In human studies, most reports using vitrified oocytes utilize ICSI (Chang et al., Reference Chang, Shapiro, Bernal, Wright, Kort and Nagy2008; Cobo et al., Reference Cobo, Castello, Vallejo, Albert, de los Santos and Remohi2013a; Kuwayama et al., Reference Kuwayama, Vajta, Kato and Leibo2005; Cao et al., Reference Cao, Xing, Li, Cong, Zhang, Wei and Zhou2009) and few have attempted IVF (Tong et al., Reference Tong, Wu, Jin, Luo, Luan and Liu2012, Reference Tong, Wu, Jin, Luan and Liu2013), giving an indication that vitrification is yet to be perfected to avoid compromising the membranes. Although, some reports have shown that increased fertilization rates can be achieved with ICSI as it can bypass the membrane barriers (Palermo et al., Reference Palermo, Joris, Devroey and Van Steirteghem1992, Reference Palermo, Neri, Takeuchi and Rosenwaks2009). It has also been reported that this technique increases the incidence of aneuploidy (Bernardini et al., Reference Bernardini, Martini, Geraedts, Hopman, Lanteri, Conte and Capitanio1997) and impairs embryonic development (Griffiths et al., Reference Griffiths, Murdoch and Herbert2000) in fresh oocytes. It has been demonstrated that blastocyst development from vitrified-warmed buffalo oocytes fertilized by either IVF or ICSI showed similar embryo development (Liang et al., Reference Liang, Srirattana, Phermthai, Somfai, Nagai and Parnpai2012; Attanasio et al., Reference Attanasio, Boccia, Vajta, Kuwayama, Campanile, Zicarelli, Neglia and Gasparrini2010). Conversely, Lane and Gardner (Reference Lane and Gardner2001) reported that there was no significant difference in blastocyst development from vitrified-warmed oocytes when compared with unfrozen oocytes (96.3% vs 95.4% respectively). In this latter study, however, a different insemination method (zona drilling) was used for post-warmed oocytes, which possibly negating the zona hardening issue.
The reduced fertilization rate of vitrified oocytes and its relationship with cortical granule exocytosis has been widely debated. Cortical granule release following initial sperm penetration is known to prevent further sperm entry, and is related to ‘hardening’ of the zona pellucida due to the cortical granule reaction. Reports on cortical granule exocytosis following oocyte vitrification are conflicting: some have shown a reduction in the number of cortical granules in post-warmed oocytes (George and Johnson, Reference George and Johnson1993; Schalkoff et al., Reference Schalkoff, Oskowitz and Powers1989; Tian et al., Reference Tian, Yan, Yang, Zhou, Yang and Zhu2007) whilst others report normal cortical granule density and distribution (Gook et al., Reference Gook, Osborn and Johnston1993; Jones et al., Reference Jones, Van Blerkom, Davis and Toledo2004; Manna et al., Reference Manna, Rienzi, Greco, Sbracia, Rahman, Poverini, Siracusa and De Felici2001; Wood et al., Reference Wood, Whittingham and Lee1992; Tong et al., Reference Tong, Wu, Jin, Luo, Luan and Liu2012). Here, we compared fresh lamb oocytes with those that had been vitrified using two combinations of cryoprotectants, DMSO + EG and PROH + EG and warmed, to determine if zona hardening or cortical granule exocytosis may have contributed to the poor fertilization rate. Cortical granules were retained within the cytoplasm of vitrified oocytes, and also showed no evidence of zona hardening, as determined by normal zona dissolution times. Some papers reported that there is no difference in zona pellucida dissolution times for unfrozen and vitrified oocytes if in vitro matured (Tian et al., Reference Tian, Yan, Yang, Zhou, Yang and Zhu2007; Tong et al., Reference Tong, Wu, Jin, Luo, Luan and Liu2012). In this study, there was no evidence of cortical granule exocytosis in lamb oocytes. In fact, vitrified oocytes showed a brighter overall staining for FITC-conjugated Lens culinaris agglutinin (LCA) throughout the cytoplasm. Ghetler et al. (Reference Ghetler, Skutelsky, Ben Nun, Ben Dor, Amihai and Shalgi2006) reported a higher intensity of fluorescence in post-warmed oocytes stained with LCA, but they interpreted this as evidence for cortical granule exocytosis (Ghetler et al., Reference Ghetler, Skutelsky, Ben Nun, Ben Dor, Amihai and Shalgi2006). Miyara et al. (Reference Miyara, Aubriot, Glissant, Nathan, Douard, Stanovici, Herve, Dumont-Hassan, LeMeur, Cohen-Bacrie and Debey2003) reported that almost 80% of oocytes displaying cytoplasmic staining for LCA showed disorganized meiotic spindles and metaphase plates (Miyara et al., Reference Miyara, Aubriot, Glissant, Nathan, Douard, Stanovici, Herve, Dumont-Hassan, LeMeur, Cohen-Bacrie and Debey2003). Interestingly, they also found that the pattern of cortical granule abnormalities in vitrified oocytes was similar to those in immature oocytes.
FITC-conjugated Lectin culinaris agglutinin (LCA) was used for its ability to bind to α-d-mannose and α-d-glucose moieties that are abundant in cortical granules (Ducibella et al., Reference Ducibella, Anderson, Albertini, Aalberg and Rangarajan1988; Lee et al., Reference Lee, Ahuja, Gilburt and Whittingham1988). One explanation for the increase in cytoplasmic LCA staining is that the cortical granules from vitrified oocytes may have become disorganized or degenerated after vitrification. Vacuoles containing degenerated cortical granules have been reported in post-warmed oocytes (Ghetler et al., Reference Ghetler, Skutelsky, Ben Nun, Ben Dor, Amihai and Shalgi2006; Hyttel et al., Reference Hyttel, Vajta and Callesen2000). Hyttel et al. (Reference Hyttel, Vajta and Callesen2000) reported an abundance of small membrane-bound vesicles containing cortical granules in the cytoplasm 0–4 h after warming, whilst 22 h later the vacuoles containing degenerate cortical granules suggesting alteration of cell biology of post-warming oocytes (Hyttel et al., Reference Hyttel, Vajta and Callesen2000). The brighter LCA fluorescence in the cytoplasm may be caused by greater non-specific binding of LCA to mucopolysaccharide moieties on cytoplasmic components other than cortical granules that are exposed after freezing. Whilst cortical granule exocytosis and zona hardening were not apparent, it is possible that the lower fertilization rate in post-warmed oocytes could be due to intrinsic modifications to the zona pellucida that are independent of cortical granule exocytosis as a consequence of cryoprotectant treatment and/or vitrification (Wood et al., Reference Wood, Whittingham and Lee1992). Partial release of cortical granules that cannot be detected by LCA staining might also have occurred (Manna et al., Reference Manna, Rienzi, Greco, Sbracia, Rahman, Poverini, Siracusa and De Felici2001).
This paper provides evidence that the effects of different cryoprotectants for oocyte vitrification in different species are not uniform. Limited success was obtained using EG alone or PROH + EG for mouse oocyte vitrification compared with DMSO + EG. Lamb oocytes demonstrated very poor fertilization and embryo development post-vitrification, and high parthenogenetic activation rates after treatment with all cryoprotectants, highlighting both the sensitivity of lamb oocytes to chilling injury and cryoprotectant toxicity during the vitrification process. There was no evidence to suggest that cortical granule exocytosis or zona hardening contributed to the poor fertilization rate of lamb oocytes. This study demonstrated a clear difference in the susceptibility of mouse and lamb oocytes to the cytotoxic effects of cryoprotectants used in the vitrification procedure. Cryoprotectant combinations therefore need to be extensively tested and carefully selected for the species being cryopreserved to optimize success rates.
Acknowledgements
We thank Castricum Brothers Abattoir for the provision of lamb ovaries and the Faculty of Veterinary Science, University of Sydney, NSW, Australia for the kind donation of lamb sperm.
Financial support
This research received no specific grant from any funding agency, commercial or non-for-profit sectors. J.S was financially supported by Australian Partnership Scholarship (APS).
Statement of interest
None.
Ethics
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional guides on the care and use of laboratory animals. All experiments using mice were approved by the Monash University Animal Ethics Committee.