Introduction
The technique of somatic cell nuclear transfer (SCNT), in which a somatic cell nucleus is transferred into an oocyte, and then reprogrammed into a pluripotent state, can be used to produce embryonic stem (ES) cells, even cloned animals (Wilmut et al., Reference Wilmut, Schnieke, McWhir, Kind and Campbell1997; Wakayama et al., Reference Wakayama, Tabar, Rodriguez, Perry, Studer and Mombaerts2001). But the application of this technique is limited due to the shortage of oocytes for certain species. Inter-species somatic cell nuclear transfer (iSCNT), an alternative SCNT technique in which a donor somatic cell from one species is transferred into a recipient oocyte of another species, may offer an opportunity to produce ES cells, and increase the population size of endangered mammals whose oocytes are extremely difficult, or sometimes impossible, to obtain (Lanza et al., Reference Lanza, Cibelli, Diaz, Moraes, Farin, Farin, Hammer, West and Damiani2000; Loi et al., Reference Loi, Ptak, Barboni, Fulka, Cappai and Clinton2001; Chen et al., Reference Chen, He, Liu, Wang, Mao, Chu, Lu, Fang, Shi, Yang, Chen, Wang, Li, Huang, Kong, Shi, Wang, Xia, Long, Xue, Ding and Sheng2003). The overall efficiency of the SCNT procedure is low (Wilmut et al., Reference Wilmut, Beaujean, de Sousa, Dinnyes, King, Paterson, Wells and Young2002; Fulka & Fulka, Reference Fulka and Fulka2007), and is even lower for iSCNT (Lee et al., Reference Lee, Kim, Park, Jeong, Lee, Park, Choi, Kim, Jeong, Kim, Hyun and Hwang2008; Lorthongpanich et al., Reference Lorthongpanich, Laowtammathron, Chan, Ketudat-Cairns and Parnpai2008; Li et al., Reference Li, Dai, Du, Zhao, Wang, Wang, Li, Liu, Wan and Li2006). It has been proposed that low cloning efficiency may be attributed largely to incomplete epigenetic reprogramming of somatic nuclei (Armstrong et al., Reference Armstrong, Lako, Dean and Stojkovic2006; Niemann et al., Reference Niemann, Tian, King and Lee2008).
Ectopic expression of certain types of transcription factors in somatic cells is an alternative technique that can be used to reprogram somatic cells into induced pluripotent stem (iPS) cells (Takahashi & Yamanaka, Reference Takahashi and Yamanaka2006; Takahashi et al., Reference Takahashi, Tanabe, Ohnuki, Narita, Ichisaka, Tomoda and Yamanaka2007; Yu et al., Reference Yu, Vodyanik, Smuga-Otto, Antosiewicz-Bourget, Frane, Tian, Nie, Jonsdottir, Ruotti, Stewart, Slukvin and Thomson2007). Recently, two studies have shown that the reprogramming of murine fibroblasts into iPS cells follows a defined sequence of molecular events that begins with the downregulation of somatic markers, followed by the upregulation of stage-specific embryonic antigen 1 (SSEA-1). SSEA-1-positive cells then gradually reactivate other markers that are associated with pluripotency, including Oct4, Nanog, Sox2, telomerase, and the silent X chromosome in female fibroblasts (Brambrink et al., Reference Brambrink, Foreman, Welstead, Lengner, Wernig, Suh and Jaenisch2008; Stadtfeld et al., Reference Stadtfeld, Maherali, Breault and Hochedlinger2008). These studies lead us consider that the low developmental ability of cloned embryos may be due to incomplete reactivation of pluripotency genes. This idea has been confirmed by recent studies, in which the expression of some pluripotency genes was monitored during the early development of cloned embryos. Most of these studies have shown that pluripotency genes, including Oct4, Sox2, and Nanog, can be reactivated in cloned embryos, although the expression level of certain pluripotency genes is somewhat abnormal (Li et al., Reference Li, Kato and Tsunoda2005; Beyhan et al., Reference Beyhan, Forsberg, Eilertsen, Kent-First and First2007; Xing et al., Reference Xing, Magnani, Lee, Wang, Cabot and Machaty2009; Aston et al., Reference Aston, Li, Hicks, Sessions, Davis, Rickords, Stevens and White2010). Interspecies cloned embryos, even offspring, have been successfully produced by iSCNT (Lanza et al., Reference Lanza, Cibelli, Diaz, Moraes, Farin, Farin, Hammer, West and Damiani2000; Loi et al., Reference Loi, Ptak, Barboni, Fulka, Cappai and Clinton2001; Murakami et al., Reference Murakami, Otoi, Wongsrikeao, Agung, Sambuu and Suzuki2005; Lorthongpanich et al., Reference Lorthongpanich, Laowtammathron, Chan, Ketudat-Cairns and Parnpai2008; Sugawara et al., Reference Sugawara, Sugimura, Hoshino and Sato2009; Hong et al., Reference Hong, Oh, Park, Kim, Kim, Koo, Jang and Lee2012; Srirattana et al., Reference Srirattana, Imsoonthornruksa, Laowtammathron, Sangmalee, Tunwattana, Thongprapai, Chaimongkol, Ketudat-Cairns and Parnpai2012), and whether pluripotency genes could be reactivated in iSCNT-derived embryos has been monitored in some studies (Chung et al., Reference Chung, Bishop, Treff, Walker, Sandler, Becker, Klimanskaya, Wun, Dunn, Hall, Su, Lu, Maserati, Choi, Scott, Atala, Dittman and Lanza2009; Wang et al., Reference Wang, Out, Chen, Lee, Latham and Cibelli2011; Hosseini et al., Reference Hosseini, Hajian, Forouzanfar, Moulavi, Abedi, Asgari, Tanhaei, Abbasi, Jafarpour, Ostadhosseini, Karamali, Karbaliaie, Baharvand and Nasr-Esfahani2012). In our previous studies, goat–sheep or bovine–sheep inter-species cloned embryos were constructed, both of which could develop in vitro to the blastocyst stage (Hua et al., Reference Hua, Zhang, Song, Song, Zhang, Zhang, Zhang, Cao and Ma2008, Ma et al., Reference Ma, Yang, Hua, Cao, Li and Zhang2008a,Reference Ma, Yang, Zhang, Cao, Hua and Lib). These results indicated that sheep oocytes could dedifferentiate a foreign somatic cell nucleus from a distant genetic correlated species. However, in these studies, whether pluripotency genes could be reactivated and their relationship with the developmental ability of iSCNT-derived embryos were not evaluated.
In the present study, in vitro matured sheep oocytes were used as nuclear recipients and sheep fetal fibroblasts (SFFs) or goat fetal fibroblasts (GFFs) were used as nuclear donors to construct sheep–sheep intra-species or goat–sheep inter-species cloned embryos. Sheep in vitro fertilized embryos were also constructed. These three types of embryos were cultured in vitro, and their developmental ability and its relationship to the reactivation of certain types of pluripotency genes were evaluated.
Materials and methods
Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA).
Donor cell preparation
SFFs or GFFs were isolated from a Mongolian sheep (ovis aries) fetus or an Guanzhong diary goat (Capra hircus) fetus, respectively, obtained from a slaughterhouse and used as nuclear donors to reconstruct sheep–sheep intra-species or goat–sheep inter-species cloned embryos. Primary cell culture, as well as freeze and thaw, were performed as described previously (Ma et al., Reference Ma, Yang, Hua, Cao, Li and Zhang2008a,Reference Ma, Yang, Zhang, Cao, Hua and Lib). Briefly, after removal of the head and internal organs, the remaining tissues were dissociated mechanically. Explants were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 20% (v/v) fetal bovine serum (FBS, HyClone, Logan, UT, USA), 100 IU/ml penicillin and 100 μg/ml streptomycin at 38°C in a humidified atmosphere of 5% CO2. When the cells from the explants reached 70% confluency, they were removed by treatment with 0.05% (quality/volume percentage concentration; m/v) trypsin–EDTA, counted, and frozen in aliquots in 10% (v/v) dimethyl sulphoxide (DMSO) and 90% (v/v) fetal bovine serum (FBS). Thawed SFFs and GFFs were plated into 96-well plates and cultured in DMEM supplemented with 10% (v/v) FBS at 38°C in a humidified atmosphere of 5% CO2 until they reached 100% confluency. The medium was then replaced with low serum medium (DMEM supplemented with 0.5% (v/v) FBS) to starve the donor cells for 3–5 days until the day of nuclear transfer (NT). Just prior to NT, the donor cells were collected by trypsinization using 0.05% (m/v) trypsin–EDTA, washed twice, and resuspended in HEPES-buffered synthetic oviduct fluid (H-SOF) supplemented with 10% (v/v) FBS.
Sheep oocyte collection and maturation in vitro
The procedure of sheep oocyte collection and maturation has been described previously (Ma et al., Reference Ma, Yang, Hua, Cao, Li and Zhang2008a,Reference Ma, Yang, Zhang, Cao, Hua and Lib). Briefly, slaughterhouse-derived ovaries were collected from mature sheep (O. aries), placed in saline (38°C) and transported to the laboratory within 1–2 h. Ovaries were washed three times in fresh phosphate-buffered saline (PBS), sliced using a microblade and the contents released in sterile Petri dishes that contained fresh PBS medium supplemented with 5% (v/v) FBS and 0.054 mg/ml heparin sodium salt. Cumulus–oocyte complexes (COCs) with several intact cumulus cell layers and a homogeneous cytoplasm were selected for maturation in vitro. COCs were washed several times, then cultured in oocyte maturation (OM) medium at 38.5°C in a humidified atmosphere of 5% CO2. After 22–24 h of culture, cumulus cells were removed from matured oocytes by vortexing the COCs for 3–5 min in Ca2+-/Mg2+-free PBS that contained 0.2% (w/v) hyaluronidase. Denuded oocytes with a polar body were selected and placed in H-SOF supplemented with 10% (v/v) FBS and prepared for enucleation.
Nuclear transfer, electrofusion, activation and embryos culture
Denuded oocytes with a polar body were incubated in H-SOF supplemented with 7.5 μg/ml cytochalasin B, 10 μg/ml Hoechst 33342 stain and 10% (v/v) FBS at 38.5°C for 15 min, then mounted onto a micromanipulator (NT-88NE; Nikon-Narishige, Tokyo, Japan) equipped with epifluorescence. Each oocyte was held with a holding micropipette (20–30 μm inner diameter, 100–150 μm outer diameter), the first polar body and adjacent cytoplasm that contained MII chromosomes were removed using an aspiration micropipette (15–18 μm inner diameter, 20–25 μm outer diameter). The removed cytoplasm was checked for the presence of chromosomes and polar bodies by exposure to UV light. Only oocytes from which all chromosomes were removed were used for NT. One of the SFFs or GFFs was gently aspirated into a micropipette and deposited in the perivitelline space of an enucleated sheep oocyte. The karyoplast–cytoplast couplets were equilibrated in an electrofused medium composed of 0.3 M mannitol, 0.5 mM HEPES, 1% (m/v) fatty acid-free BSA (FAFBSA), 0.05 mM CaCl2 and 0.1 mM MgCl2 for 3 min, then transferred into a cell fusion chamber that contained the same medium used for electrofusion and using a fusion machine (EP-1 Voltain, CryoLogic Pty Ltd, Melbourne, Australia). Karyoplast–cytoplast couplets were aligned manually, then subjected to a double DC fusion pulse of 1.25 kV/cm for 80 μs, as described previously (Beaujean et al., Reference Beaujean, Taylor, Gardner, Wilmut, Meehan and Young2004). After electrofusion, the karyoplast–cytoplast couplets were transferred into H-SOF supplemented with 10% (v/v) FBS whilst waiting for fusion to complete. The non-fused couplets were subjected to a second round of fusion 1 h after the first. The fused embryos were activated by culture in H-SOF that contained 5 μM ionomycin and 10% (v/v) FBS for 5 min and were subsequently cultured in SOF that contained 2 mM 6-DMAP and 10% (v/v) FBS for 4 h. The activated embryos were washed twice and cultured in SOF supplemented with 2% (v/v) essential amino acids (Gibco); 1% (v/v) non-essential amino acids (Gibco); 8 mg/ml FAFBSA, 5% (v/v) FBS and 1 mM glutamine as described previously (Loi et al., Reference Loi, Ptak, Barboni, Fulka, Cappai and Clinton2001). The embryos were monitored every 24 h for the progression of development and half the culture medium was replaced with fresh medium every 48 h.
In vitro fertilization and embryos culture
The procedure of in vitro fertilization has been described previously and was followed with some modification (Borowczyk et al., Reference Borowczyk, Caton, Redmer, Bilski, Weigl, Vonnahme, Borowicz, Kirsch, Kraft, Reynolds and Grazul-Bilska2006). Frozen rams semen was thawed in 2 ml of fertilization medium composed of SOF supplemented with 20% oestrus sheep serum, 5 IU/ml heparin, 5 mM caffine, and were kept in a CO2 incubator (38.5°C) at an angle of 45°. The sperm were allowed to swim up for 30 min. The matured oocytes were washed 2–3 times and then transferred to 2 ml of fertilization medium. The highly motile spermatozoa from the upper layers were added to the oocytes at a concentration of 1–2 × 106/ml approximately, and incubated for 18 h at 38.5°C in a humidified atmosphere of 5% CO2. Then, the presumptive fertilized embryos were washed 2–3 times and cultured in modified SOF as described above; the progression of embryo development was monitored every 24 h and half the culture medium was replaced every 48 h.
Immunohistochemical labeling of SSEA-1 on the surface of embryo
Embryos derived from nuclear transfer and in vitro fertilization were collected at the different developmental stages, 2-cell, 4-cell, 8-cell and morula. Embryos were washed twice in PBS, then placed in PBS supplemented with 0.25% (m/v) pronase for 3–5 min. The zona pellucida was removed completely, then embryos were transferred immediately into PBS. Mouse anti-SSEA-1 monoclonal antibody (Developmental Studies Hybridoma Bank, Iowa City, IA, USA) was used to detect SSEA-1 on the surface of embryos as described previously (Kühholzer et al., Reference Kühholzer, Baguisi and Overström2000; Yan et al., Reference Yan, Lei, Yang, Gao, Lei, Ma and Dou2008; Park et al., Reference Park, Colletti, Ozturk, Wood, Tellez, Almeida-Porada and Porada2009). In brief, embryos were incubated in mouse anti-SSEA-1 monoclonal antibody (1:100) at room temperature for 30 min. Embryos were washed twice in PBS, then incubated in a secondary antibody solution (1.5 μg/ml), either FITC- or TRITC-conjugated rabbit anti-mouse IgG + IgM (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 30 min at room temperature in the dark. Subsequently, embryos were washed once in PBS, then placed in a microdroplet of PBS, overlaid with paraffin oil, and observed under an epifluorescence microscope.
Collection of total RNA from embryos
Embryos derived from nuclear transfer and in vitro fertilization were collected at different developmental stages, 2-cell, 4-cell, 8-cell, morula and blastocyst, and washed three times in PBS. Then, individual embryos were transferred into polymerase chain reaction (PCR) tubes that contained 18 μl ice-cold Cells-to-cDNA II Cell Lysis Buffer (Ambion, Applied Biosystems Inc., Foster City, CA, USA). The mixture was incubated at 75°C for 15 min, then removed from the heat source and placed on ice. Next, 2 μl RNase-free DNase I (2 U/μl, Ambion) was added and the mixture was incubated at 37°C for 15 min to degrade genomic DNA, and then subsequently incubated at 75°C for 5 min to inactivate DNase I, centrifuged and then used as a template for reverse transcription PCR (RT-PCR). In addition, total RNA was extracted from SFFs and GFFs using the same procedure as described above.
RT-PCR and products sequencing
Four pairs of RT-PCR primers were designed based on the sequences of sheep or goat Oct4 mRNA, or sheep or goat Nanog mRNA, using Primer Premier 5.0 software and described in Table 1. In addition, details of positive control RNA from the TaKaRa One Step RNA PCR Kit (TaKaRa, Dalian, Liaoning, China) and its corresponding primers are listed in Table 1. Total RNA from embryos was subjected to RT-PCR using the TaKaRa The One Step RNA PCR Kit. RT-PCR reaction mixture consisted of 5 μl 10× One Step RNA PCR buffer, 10 μl MgCl2 (25 mM), 5 μl dNTP mixture (10 mM each), 1 μl RNase inhibitor (40 U/μl), 1 μl AMV RTase XL (5 U/μl), 1 μl AMV-Optimized Taq (5 U/μl); 1 μl forward primer (20 μM), 1 μl reverse primer (20 μM), 1 μl positive control forward primer (20 μM), 1 μl positive control reverse primer (20 μM), 20 μl total RNA, 1 μl positive control RNA and 2 μl RNase-free H2O in a total volume of 50 μl. Moreover, total RNA extracted from SFFs and GFFs was also subjected to RT-PCR in the same reaction system as described above, and was used as a negative control to confirm that the total RNA from embryos and somatic cells was not contaminated by genomic DNA. An RT reaction was carried out at 42°C for 30 min, followed by a step of 94°C for 2 min to inactivate AMV RTase. The RT reaction mixture was used directly for PCR. PCR was performed with the parameters of denaturation at 94°C for 30 s, annealing at a specified temperature (45°C for sheep-specific primers and 50°C for goat-specific primers) for 30 s and an extension step at 72°C for 60 s using a thermocycler (DNA Engine DYAD; MJ Research Inc., Waltham, MA, USA). After 45 cycles, the correct PCR products were identified by electrophoresis and sequencing.
Table 1 Information on the primers used for RT-PCR

Statistical analyses
The developmental rate of embryos was compared statistically by chi-squared analysis using SPSS software (SPSS Inc., Chicago, IL, USA). Differences at a P-value <0.05 were considered to be statistically significant.
Results
Developmental ability of embryos
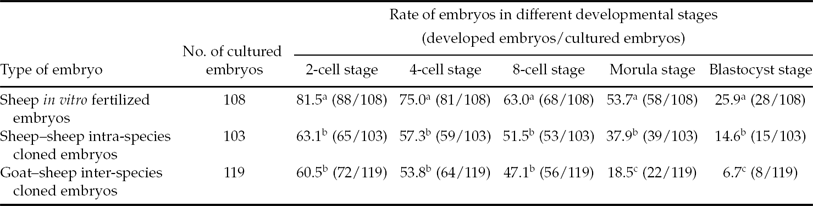
After culture in vitro, all types of embryos were able to develop into blastocysts, even hatched blastocysts (Fig. 1), but the developmental rates were different. As shown in Table 2, at different developmental stages, the developmental rate of in vitro fertilized embryos was significantly higher than that of cloned embryos. At the 2-cell to 8-cell stages, the two types of cloned embryos had comparable developmental ability. However, at the morula and blastocyst stages, the developmental rate of intra-species cloned embryos was significantly higher than that of the inter-species cloned embryos.
Table 2 The development of embryos derived from in vitro fertilization and nuclear transfer*
Within the same column, values with different superscripts are significantly different (P < 0.05).
*Data were collected from 15 series of nuclear transfer and in vitro fertilization experiments.
Figure 1 The development of embryos derived from in vitro fertilization and nuclear transfer. (A1) blastocyst derived from intra-species nuclear transfer. (A2) Hatched blastocyst derived from intra-species nuclear transfer. (B1) Blastocyst derived from inter-species nuclear transfer. (B2) Hatched blastocyst derived from inter-species nuclear transfer. (C1) Blastocyst derived from in vitro fertilization. (C2) Expanded blastocyst derived from in vitro fertilization.
Expression of SSEA-1 on the surface of embryos
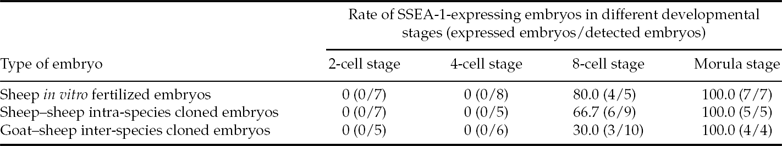
As shown in Fig. 2, SSEA-1 could be detected on the blastomere's outer surface in cloned or in in vitro fertilized embryos at the 8-cell and morula developmental stages. Interestingly, as shown in Table 3, not all embryos could express SSEA-1 at the 8-cell stage; only 80.0, 66.7 and 30.0% of in vitro fertilized embryos, intra-species and inter-species cloned embryos, respectively, could express SSEA-1. However, at the morula stage, all detected embryos could express SSEA-1.
Figure 2 Detection of SSEA-1 on the surface of embryos. (A, B) mouse anti-SSEA-1 monoclonal antibody was used as primary antibody, FITC-conjugated rabbit anti-mouse IgG + IgM was used as secondary antibody, SSEA-1 on the surface of intra-species cloned embryos were detected. (C, D) mouse anti-SSEA-1 monoclonal antibody was used as primary antibody, TRITC-conjugated rabbit anti-mouse IgG + IgM was used as secondary antibody, SSEA-1 on the surface of goat–sheep inter-species cloned embryos were detected. (E, F) Mouse anti-SSEA-1 monoclonal antibody was used as primary antibody, FITC-conjugated rabbit anti-mouse IgG + IgM was used as secondary antibody, SSEA-1 on the surface of in vitro fertilized embryos were detected. (A, C, E) Observed under visible light; (B, D, F) observed under fluorescence. (A1–F1): 2-cell embryos; (A2–F2): 4-cell embryos; (A3–F3): 8-cell embryos; (A4–F4) morula.
Table 3 The expression of SSEA-1 on the surface of embryos derived from in vitro fertilization and nuclear transfer*
*Data were collected from three series of immunohistochemical labeling experiments.
Expression of Oct4 and Nanog mRNA in embryos
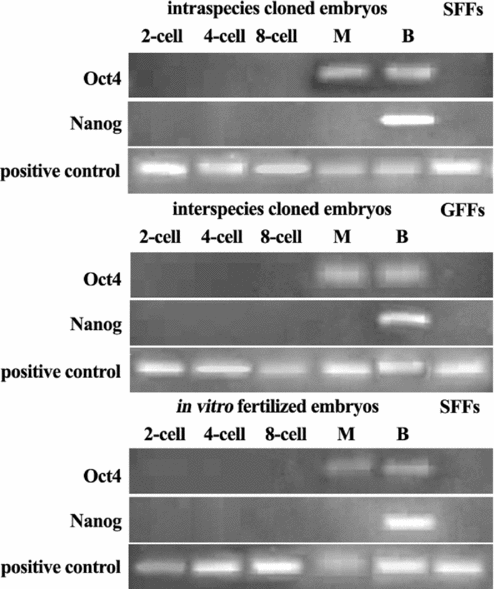
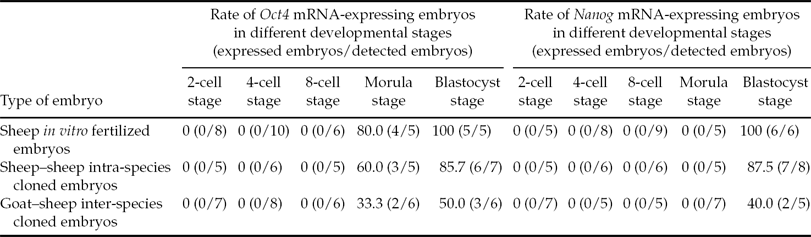
As shown in Fig. 3, all types of embryos expressed Oct4 mRNA at the morula (M) and blastocyst (B) stages. Moreover, as shown in Table 4, at the morula stage only 80.0, 60.0 and 33.3% of in vitro fertilized embryos, intra-species and inter-species cloned embryos, respectively, could express Oct4 mRNA. At the blastocyst stage, the rate of Oct4 mRNA-expressing embryos increased to 100, 85.7 and 50.0% respectively for three types of embryos. Also shown in Fig. 3, Nanog mRNA was expressed in embryos as late as the blastocyst stage; the rate of Nanog mRNA-expressing embryos was 100, 87.5 and 40.0% respectively for three types (Table 4).
Figure 3 Electrophoresis of Oct4 and Nanog cDNA.
Table 4 The expression of Oct4 and Nanog mRNA in embryos derived from in vitro fertilization and nuclear transfer*
*Data were collected from 5–10 series of RT-PCR experiments.
When total RNA extracted from SFFs or GFFs was used as a template, Oct4 and Nanog mRNA products could not be amplified by RT-PCR (Fig. 3). This result indicated that RT-PCR products were derived from mRNA, not from genomic DNA. Moreover, products of positive control RNA could be amplified in all RT-PCR reactions (Fig. 3), this indicated that the RT-PCR reaction system was applicable for use. The sizes of the RT-PCR products are shown in Table 1, and were confirmed by electrophoresis (Fig. 3) and sequencing (data not shown).
Discussion
In present study, embryos with the lowest developmental ability were inter-species cloned embryos, as only 6.7% of fused embryos could develop to the blastocyst stage, and this rate was significantly lower than that of in vitro fertilized embryos (25.9%) and intra-species cloned embryos (14.6%). This result indicated that sheep oocytes had the capacity comparatively to reprogram homogenous donor nuclei. Similar results have been obtained in other studies, in which bovine and rabbit oocytes were used as nuclear recipients, and in which the developmental ability of intra-species cloned embryos was significantly higher than that of inter-species cloned embryos (Murakami et al., Reference Murakami, Otoi, Wongsrikeao, Agung, Sambuu and Suzuki2005; Li et al., Reference Li, Dai, Du, Zhao, Wang, Wang, Li, Liu, Wan and Li2006; Zhao et al., Reference Zhao, Ouyang, Nan, Lei, Song, Sun and Chen2006; Lorthongpanich, et al., Reference Lorthongpanich, Laowtammathron, Chan, Ketudat-Cairns and Parnpai2008).
In the present study, the developmental progression of cloned embryos showed that the difference in blastocyst rate between intra-species and inter-species cloned embryos was mainly due to poor development of inter-species cloned embryos from the 8-cell to the morula stages. Zygotic gene activation (ZGA) is the critical event that governs the transition from maternal to embryonic control of development, and a failure in ZGA can result in the developmental block of embryos. Previous studies have shown that, in both sheep and goat embryos, ZGA occurs at the 8-cell to 16-cell stages (Ferrer et al., Reference Ferrer, Garcia, Villar and Arias1995; Pivko et al., Reference Pivko, Grafenau and Kopecný1995). Therefore, in the present study, the low blastocyst rate of inter-species cloned embryos is largely attributed to a failure in ZGA. This conclusion was confirmed as only 30% of inter-species cloned embryos could reactivate gene of SSEA-1 at the 8-cell stage, a figure that was lower than that of in vitro fertilized embryos (80.0%) and intra-species cloned embryos (66.7%).
SSEA-1 is a type of ES (or ES-like) cell surface marker in several species, including mouse, rat, goat, and sheep (Solter & Knowles, Reference Solter and Knowles1978; Vassilieva et al., Reference Vassilieva, Guan, Pich and Wobus2000; He et al., Reference He, Pant, Schiffmacher, Bischoff, Melican, Gavin and Keefer2006; Dattena et al., Reference Dattena, Chessa, Lacerenza, Accardo, Pilichi, Mara, Chessa, Vincenti and Cappai2006). During early development of mouse embryos, SSEA-1 is first detected on blastomeres at the 8-cell stage (Solter & Knowles, Reference Solter and Knowles1978), whereas in other species the early expression of SSEA-1 has not been reported. In the present study, three types of embryos initially expressed SSEA-1 at the 8-cell stage, and SSEA-1 was still expressed at the morula stage. This result indicated that the timing of SSEA-1 expression is identical during early development of mouse, sheep and goat embryos. Moreover, in the present study, a low percentage of intra-species cloned embryos could express SSEA-1 at the 8-cell stage compared with in vitro fertilized embryos, this rate was even lower in inter-species cloned embryos. We speculate that this result may be a consequence of only small numbers of cloned embryos able to successfully reactivate zygotic genes, which subsequently resulted in a lower rate of SSEA-1-expressing embryos in cloned embryos. Furthermore, statistical analysis showed that, at the 8-cell stage, the rate (80.0% for in vitro fertilized embryos; 66.7% for intra-species and 30.0% for inter-species cloned embryos; Table 3) of SSEA-1-positive embryos was roughly comparable with the developmental rate (85.3% for in vitro fertilized embryos; 73.6% for intra-species and 39.3% for inter-species cloned embryos; Table 2) of 8-cell to morula stages, although the former was somewhat lower than the latter. This difference could be because the detection of SSEA-1 was performed before the occurrence of ZGA in some embryos. Therefore, SSEA-1 could be used as a surface marker to estimate the percentage of embryos that have already successfully reactivated zygotic genes and have the ability to develop into morula.
Oct4 and Nanog are transcription factors that are required to maintain the pluripotency and self-renewal of ES cells (Loh et al., Reference Loh, Wu, Chew, Vega, Zhang, Chen, Bourque, George, Leong, Liu, Wong, Sung, Lee, Zhao, Chiu, Lipovich, Kuznetsov, Robson, Stanton, Wei, Ruan, Lim and Ng2006). During the early development of mammal embryos, Oct4 and Nanog can be expressed at certain stages. In mouse embryos, the initial expression of Oct4 and Nanog is at the 8-cell and morula stages respectively, whereas in bovine embryos, these two genes are initially expressed at the 16-cell and blastocyst stages respectively (Chambers et al., Reference Chambers, Colby, Robertson, Nichols, Lee, Tweedie and Smith2003; Kurosaka et al., Reference Kurosaka, Eckardt and McLaughlin2004). In the present study, three types of embryos initially expressed Oct4 and Nanog mRNA at the morula and blastocyst stages respectively. These results indicated that the expression pattern of Oct4 and Nanog mRNA is similar in ruminants. Moreover, in the present study, the failure to reactivate Oct4 and Nanog genes was observed in some embryos, although these embryos could develop to the morula stage, even the blastocyst stage, and this phenomenon more frequently occurred in inter-species cloned embryos. In previous studies, the failure to reactivate pluripotency genes and their abnormal expression were also observed in a some intra-species and inter-species cloned embryos (Boiani et al., Reference Boiani, Eckardt, Schöler and McLaughlin2002; Chung et al., Reference Chung, Bishop, Treff, Walker, Sandler, Becker, Klimanskaya, Wun, Dunn, Hall, Su, Lu, Maserati, Choi, Scott, Atala, Dittman and Lanza2009; Wang et al., Reference Wang, Out, Chen, Lee, Latham and Cibelli2011). These studies, together with our study, indicated that the developmental competence of some cloned embryos is already compromised at the blastocyst stage due to non-activation of certain pluripotency genes. This factor is possibly a reason for the high pregnancy loss of cloned embryos and low survival of cloned offspring.
In conclusion, three types of embryos can develop in vitro to the blastocyst stage. In vitro fertilized embryos have a higher blastocyst rate than cloned embryos, and ZGA has an important influence on the blastocyst rate of cloned embryos. Moreover, only some cloned embryos can express SSEA-1, Oct4 and Nanog genes during early development; the failure in reactivation of some pluripotency genes may be a reason for the low survival rate of cloned embryos in subsequent development.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 31160245); Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region; Natural Science Foundation of Inner Mongolia Autonomous Region of China (grant nos. 2009BS0503, 2012MS0503); Research Program of Natural Science at Universities of Inner Mongolia Autonomous Region of China (grant no. NJ09093) and Innovation Foundation of Inner Mongolia University of Science & Technology (grant nos. 2009NC058, 2011NCL007).