Introduction
The Indian river buffalo (Bubalus bubalis) is a major dairy animal in India that produces about two-thirds of the world's buffalo milk and nearly half of the world's buffalo meat (FAOSTAT, 2005). However, these animals suffer from obvious disadvantages that make them slow reproducers (Nandi et al., Reference Nandi, Raghu, Ravindranatha and Chauhan2002). As an aid to augment reproduction rate in buffalo, numerous assisted reproduction technologies (ARTs) have been tried with limited success (Gasparrini, Reference Gasparrini2002; Gasparrini et al., Reference Gasparrini, Boccia, Marchandise, Di Palo, George, Donnay and Zicarelli2006, Drost, Reference Drost2007). Information on expression pattern regulation of oocyte-expressed genes is crucial for deciding strategies on how the oocytes could be stimulated to gain optimum development ability, which would ultimately affect the health of resulting embryos and ultimately the success rate of ART protocols in this species (Sirard et al., Reference Sirard, Richard, Blondin and Robert2006).
Growth differentiation factor 9 (GDF9) plays an important role in bi-directional communication between oocytes and its surrounding cumulus cells to provide developmental competence to oocytes (Matzuk et al., Reference Matzuk, Burns, Vivieros and Eppig2002). GDF9 is expressed in oocytes throughout most stages of folliculogenesis and persists even after fertilization, through preimplantation embryo development (McGrath et al., Reference McGrath, Esquela and Lee1995). Modulation of expression patterns of developmentally important genes during in vitro maturation and fertilization has been cited as an important reason for the compromised developmental competence of oocytes and embryos produced through ARTs (Knijin et al., Reference Knijin, Wrenzycki, Hendriksen, Vos, Hermann, Vander, Niemann and Dielemen2002).
In spite of the importance of GDF9, however, information regarding the structure of its regulatory regions has started to emerge only recently. Elucidating the cDNA structure and regulatory elements of this gene will help in the understanding the mechanism of its oocyte-specific expression control during different phases of follicular growth. Such information will also help in production of buffalo-specific recombinant GDF9 for therapeutic applications (Hayashi et al., Reference Hayashi, Mcgee, Min, Klein, Rose, Duin and Hsueh1999). Analysis of the nucleotide and predicted amino acid sequences of the GDF9 gene can provide further insight into the evolution of this family of genes (TGF-β). The cDNA sequence of GDF9 has been characterized in mice, bovine and porcine (Incerti et al., Reference Incerti, Dong, Borsani and Matzuk1994; Sendai et al., Reference Sendai, Itoh, Yamashita and Hoshi2001; Shimizu et al., Reference Shimizu, Miyahayashi, Yokoo, Hoshino, Sasada and Sato2004). In buffaloes, however, no information is available except partial cDNA sequence reported earlier from our laboratory (NCBI GenBank accession no. EF202171). In the present study GDF9 cDNA prepared from buffalo oocyte RNA has been sequenced and characterized. To further understand the control of GDF9 transcription, we characterized the transcription start site (TSS) of this gene. The about 5 kb 5′ upstream region of the buffalo GDF9 gene was amplified and the derived sequence was analyzed to define putative cis-acting regulatory elements that may be responsible for oocyte-specific expression of GDF9.
Materials and methods
Oocyte collection
Buffalo ovaries were collected from an abattoir and transported to the laboratory in pre-warmed normal saline (35–37°C) supplemented with antibiotics (streptomycin sulphate 100 μg/ml and penicillin 100 U/ml). Cumulus–oocyte complexes (COCs) were collected within 4–6 h after slaughter of animals by aspiration from large (≥6) and small (≤3) follicles by using a 18G needle attached to a 10 ml syringe in oocyte collection medium (Dulbecco's phosphate-buffered saline (PBS) supplemented with 0.01% l-glutamine, 0.4% bovine serum albumin (BSA), 100 μg/ml streptomycin sulphate, 100 U/ml penicillin and sodium pyruvate 36 μg/ml). COCs were selected according to their morphological characteristics and graded on the basis of presence of number of cumulus layers and homogeneity of ooplasm. Only excellent grade oocytes with more than five layers of cumulus mass were selected for in vitro maturation.
Oocyte maturation
A pool of 25 COCs was put for in vitro maturation in maturation medium (TCM-199 with 10% fetal bovine serum (FBS), 0.005% streptomycin, 0.01% sodium pyruvate and 0.005% glutamine) supplemented with 5 μg/ml each of follicle stimulating hormone (FSH; porcine, Sigma) and luteinizing hormone (LH; Sigma) and 50 ng/ml epidermal growth factor (EGF; Sigma). Maturation drops (100 μl) were prepared in advance and overlaid with autoclaved mineral oil and equilibrated for at least 2 h before addition of the oocytes. For in vitro maturation, oocytes placed in maturation drops were incubated in CO2 incubator at 38.5°C in a 5% CO2 in air atmosphere. COCs were taken out from maturation drops after 8 h of culture and washed with 1× PBS. Washed COCs were then denuded by vortexing for 3 min and taken in an RNase-free Eppendorf tube in minimum volume of PBS after washing them 3–4 times with 1× PBS to make sure that there is no contamination with cumulus cells. A batch of 100 oocytes was kept in 50 μl of Trizol reagent (TRI Reagent™, Sigma) at –80°C until RNA isolation. Likewise the pool of oocytes representative of different follicular categories (as mentioned before) was collected for further processing.
RNA Isolation and reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated from the oocyte pool, using TRI Reagent (Sigma, USA) as per manufacturer's instruction. The RNA pellet was finally dissolved in 20 μl of RNase-free water. The quality of RNA was checked by migrating 2 μl aliquots on 1.5% formaldehyde agarose gel. RT-PCR was performed using the Superscript First Strand cDNA synthesis RT-PCR kit (Invitrogen), as per the manufacturers’ protocol. Briefly, isolated RNA was treated with RNase-free DNase to remove any contaminating genomic DNA. Next, 500 ng of total RNA was reverse transcribed using random oligoprimers. PCR analysis of GDF9 mRNA was performed using two sets of primers amplifying 730 bp (part A) 731 bp (part B) with 94 bp of overlapping sequence (Fig. 1). The primers used to amplify the GDF9 cDNA were A1 and A2 for part A and B1, and B2 for part B (Table 1). Non-reverse transcribed RNA samples were used as the negative control. RT-PCR products were fractionated by size on a 2% agarose gel and visualized by ethidium bromide (EtBr) fluorescence under ultraviolet (UV) light illumination. PCR products were purified using the QIAquick Gel extraction kit (Qiagen) and sequenced in both directions. The cDNA sequence was reconstructed by aligning the overlapping region of part A and part B.

Figure 1 Primer strategy used for GDF9 cDNA synthesis: A1, A2 for fragment A (730 bp) and B1, B2 for fragment B (731 bp); R1, R2 for 5′ RLM-RACE, F1, F2 for 3′ RACE, P1 to P10 overlapping sets of primers for amplification of 5′ flanking region of GDF9. TSS: transcriptional start site, ▼ indicates the translational start site.
Table 1 Sequence and position of the oligonucleotides used in PCR, 5′ RACE PCR and amplification of the 5′ flanking region of buffalo GDF9 gene

(+) and (-) indicate sense and antisense primers respectively, primer positions are numbered relative to the +1 translation start site (ATG)
Amplification of 5′ and 3′ cDNA ends of GDF9
The FirstChoice® RLM-RACE Kit (Ambion) was used to amplify the GDF9 5′ cDNA end, following manufacturer's instruction. Briefly, 8 μg of total RNA extracted from a pool of in vitro matured oocytes was treated with calf intestinal phosphatase to remove the free 5′-phosphate group. Tobacco acid pyrophosphatase was then used to specifically remove the cap structure from the full-length mRNA, leaving a 5′ monophosphate. Non-tobacco acid pyrophosphatase-treated RNAs were used as negative controls. A RNA oligonucleotide adaptor was ligated to the newly decapped mRNA by T4 RNA ligase. Using the ligated RNA as template, GDF9 cDNA was synthesized by reverse transcription using M-MLV reverse transcriptase and (dT) 15 primers. The resulting cDNA was then amplified by nested PCR using Accuprime (Hotstart) Taq DNA polymerase (Invitrogen) using buffalo GDF9 gene-specific primers (reverse) and the adaptor primers (forward, provided by the manufacturer). The gene-specific antisense inner primer R1 and the nested PCR outer primer R2 (Table 1) were designed for the RLM-RACE based on the buffalo GDF9 cDNA sequence derived in the present study. The 5′ RLM-RACE PCR products were analyzed on 2% agarose gels and cloned into pCR 2.1 vector for sequencing, using the TOPO-TA cloning kit (Invitrogen). For 3′ RLM-RACE, total RNA was extracted from a pool of 800 in vitro matured oocytes (2.4 μg), representing different follicular categories and reverse transcribed with the 3′ adapter supplied with the kit. The resultant cDNA was amplified with the gene-specific outer forward primer F1 (Table 1) and adaptor-specific outer reverse primer provided with the kit. A second round of nested PCR reactions was carried out by using the set of gene-specific inner forward primer F2 (Table 1) and adaptor-specific inner reverse primer. All PCR amplifications were done with Accuprime (Hotstart) Taq DNA polymerase (Invitrogen). The 3′ RACE PCR product was confirmed on 2% agarose, then extracted from the gel, purified and sequenced by standard dideoxy method.
Isolation of 5′ flanking region of buffalo GDF9 gene
A standard proteinase K digestion method followed by phenol–chloroform extraction (Sambrook & Russell, Reference Sambrook and Russell2001) was used to isolate genomic DNA from peripheral blood leukocytes for the analysis of 5′ flanking region of GDF9 gene of buffalo. A primer walking approach was used involving five sets of PCR primers (Table 1) designed for specific amplification of the 5′ flanking region of GDF9 gene by an overlapping approach (Fig. 1). As no buffalo-specific sequence was available for GDF9, DNA sequences from other related species available in GenBank (www.ncbi.nlm-nih.gov.in) were analyzed for homology alignment (using ClustalW) and primers were decided at consensus sequences and custom synthesized from Sigma Aldrich Chemical Com, Aldrich, USA. PCR was performed using Pfx DNA polymerase (Invitrogen, USA) under the following thermal condition: one cycle at 95°C for 2 min, 31 cycles at 95°C for 15 s, 58°C for 30 s and 68°C for 1 min and finally extension at 72°C for 8 min in a thermal cycler (Eppendorf, USA). All the primer sets were amplified in the same conditions and the resulting PCR products were confirmed on 2% agarose gels. The PCR products were purified using the QIAquick gel extraction kit (Qiagen, USA) and sequenced. The targeted 4244-bp sequence upstream from TSS was reconstructed by aligning the overlapping region of the five individual fragments amplified by PCR. The 5′ flanking sequence was deposited in GenBank (accession no. FJ529502).
Sequence analysis and TF (transcription factor) binding site prediction
The buffalo GDF9 amino acid sequence was deduced and comparative analysis carried out with other vertebrates GDF9 amino acid sequences using the ClustalW homology alignment program and the Bioedit version 7.0.5.3 software. Available vertebrate GDF9 amino acid sequences (from GenBank and Ensemble) considered as references were bovine (NP_777106.1), sheep (AAC28089.2), goat (ABR10699.1), pig (NP_001001909.1), dog (ENSCAFG00000000916), horse (XP_001504477.1), human (NP_005251.1), mice (NP_032136.2), rat (EDM04383.1), chicken (NP_996871.2) and zebrafish (AAI08014.1). The TSS position on buffalo GDF9 was determined by aligning the sequence of 11 randomly selected clone of RACE amplified product. The 5′ flanking regions of cow (ENSBTAG00000009478), human (ENSG00000164404) and mouse (ENSMUSG00000018238) available at Ensemble Genome Browser (http://www.ensembl.org/index.html) were used for comparative analysis of the worked out 5′ flanking region of buffalo GDF9 using the ClustalW homology alignment program.
The buffalo, cow, human and mouse 5′ flanking region was analyzed using the MatInspector program (Quandt et al., Reference Quandt, Frech, Karas, Wingender and Werner1995). For the presence of transcription factor binding sites (solution parameters: core similarity 1.0; matrix-optimized), similar but not identical germ cell-specific transcription factors binding sites were screened manually and are listed in Table 2.
Table 2 Predicted transcription factors binding sites at 5′ upstream of the buffalo GDF9 sequence

The nucleotide numbers mentioned are with reference to ATG start codon identified on buffalo GDF9 5′ upstream region. Nucleotides in capital indicate consensus core binding sites.
Results
Sequence analysis of buffalo GDF9 cDNA
The orthologous oligonucleotide primers designed from the available GDF9 cDNA sequence data for bovine and other related species amplified the expected fragments of 730 bp (Part A) and 731 bp (Part B) from the buffalo oocyte cDNA. (Fig. 1). Resultant cDNA sequence was reconstructed with 5′UTR and 3′UTR data (obtained from RACE experiments) to interpret the complete buffalo GDF9 cDNA sequence. It consists of a 1823 bp long sequence that included 57 bp of the 5′ UTR (position 1–57), 1359 bp (position 57–1416) corresponding to 453 amino acid of whole protein, which is composed of proregion (318 residues), mature protein (135 residues), a stop codon (TAA, position 1417–1420), and 404 bp of the 3′ UTR region. The deduced amino acid sequence was found to contain three potential N-glycosylation sites (112, 269, 405) and eight cysteine residues at positions 12, 176, 352, 381, 385, 418, 450 and 452 that appeared to be conserved across 12 different vertebrate species (Fig. 2). A cluster of basic amino acids in the putative cleavage site (RHRR; boxed in Fig. 3) between proregion and the mature protein was found to be conserved in buffalo along with other vertebrate species.

Figure 2 Predicted buffalo GDF9 peptide representing amino (NH2) and carboxyl (COOH) terminus; open box indicates the proregion and shaded box for mature region, N: possible N-linked glycosylation sites, C: conserved cysteine residues.

Figure 3 Homology analysis of GDF9 amino acid sequences between buffalo (FJ529501) and other vertebrates, including bovine (NP_777106.1), sheep (AAC28089.2), goat (ABR10699.1), porcine (NP_001001909.1), dog (ENSCAFG00000000916), horse (XP_001504477.1), human (NP_005251.1), mouse (NP_032136.2), rat (EDM04383.1), chicken (NP_996871.2) and zebrafish (AAI08014.1). Dashes indicate gaps for optimal alignment. Sequences conserved across all entries are marked with shadow. Boxed area represents the cleavage site between pro- and mature protein AA sequences. N-linked glycosylation sites and conserved cysteine residues are marked with ▼ and * respectively.
Mapping of transcription start site and identification of 3′UTR
The 5′ RLM-RACE product in principle was expected to reveal the TSS of GDF9, as tobacco acid pyrophosphatase (TAP) used in the RACE protocol picks up only the 5′ methyl cap intact RNA species. Figure 4 depicts at least 11 randomly selected cloned 5′ RACE products with uniform size. The repeated observation of an ~400 bp product was indicative of the presence of a constant TSS for expressed GDF9 from different follicular category oocytes. The exact TSS location was further confirmed by comparison of sequences obtained from these clones. The TSS was mapped to a position at 57 bp upstream of the ATG. Further, 3′ RACE analysis also revealed a single RACE product of ~600 bp which was further confirmed by customized sequencing (Fig. 5). With the sequencing result of this single 3′ UTR region, it was then clubbed with the identified GDF9 cDNA sequence to produce a length of 404 bp (including polyA tail) from termination codon TAA.

Figure 4 5′ RLM-RACE product of GDF9 gene in oocytes obtained from a heterogeneous pool of follicles from buffalo ovary. (A) ~400 bp primary 5′RLM-RACE amplified product (P). (B) RLM-RACE product was confirmed in multiple randomly picked clones (1–11). L: 100 bp ladder.

Figure 5 3′RLM-RACE product of GDF9 gene in oocytes. (A) ~600 bp primary 3′ RLM-RACE amplified product. (B) The RACE PCR product confirmed in multiple clones (1–3). L: 100 bp ladder.
Isolation and analysis of 5′ flanking region of the GDF9 gene
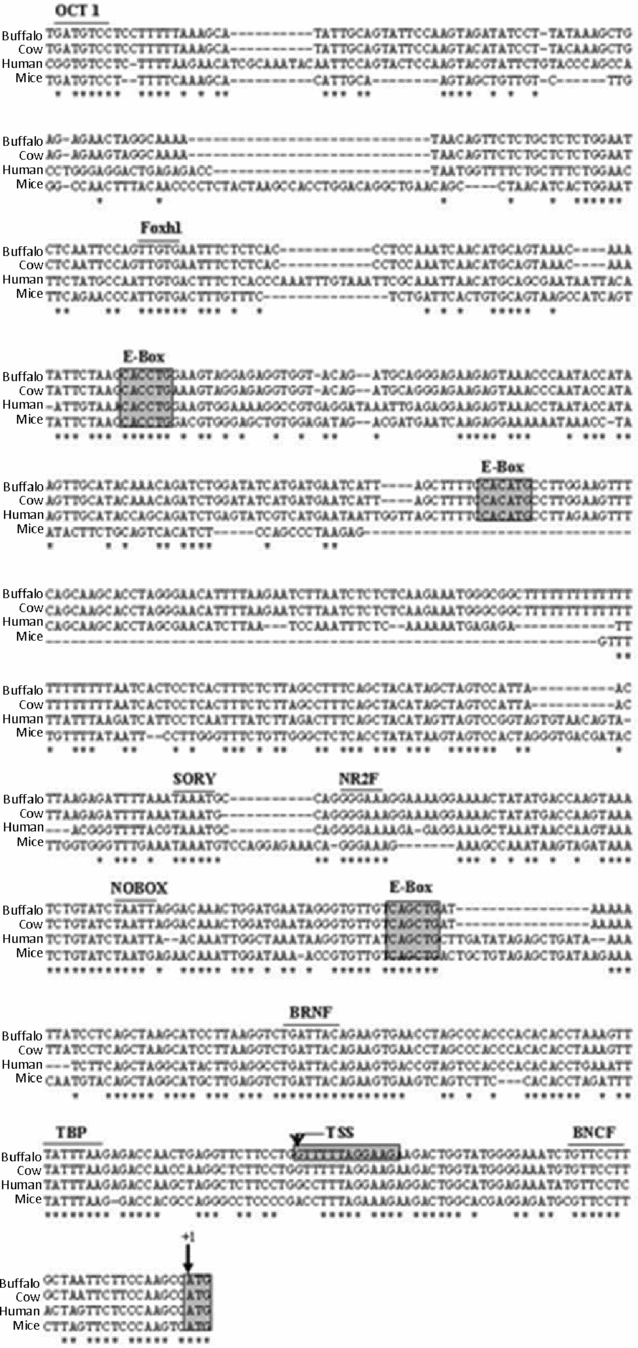
On the basis of identified TSS position, the derived buffalo GDF9 genomic DNA sequence of 4244 bp that encompassed the 5′ upstream region was aligned with the corresponding GDF9 sequence in other mammalian species such as cow, human and mouse. It was observed that the initial (to ATG) 753 bp region is highly conserved in all species, as shown in Fig. 6. This region was found to contain core promoter elements such as a TATA box at position –23 bp relative to the TSS. Other germ cell-specific TF binding sites, such as three E-box elements and a NOBOX binding element (NBE) were also found to be conserved across species (Fig. 6). The remaining 3490-bp upstream sequence was analyzed for the presence of putative germ cell-specific transcription factor binding sites. Table 2 summarizes all the predicted TFs in the 4244 bp sequence, their positions related to ATG and corresponding binding sites mapped on buffalo GDF9 5′ upstream sequence.
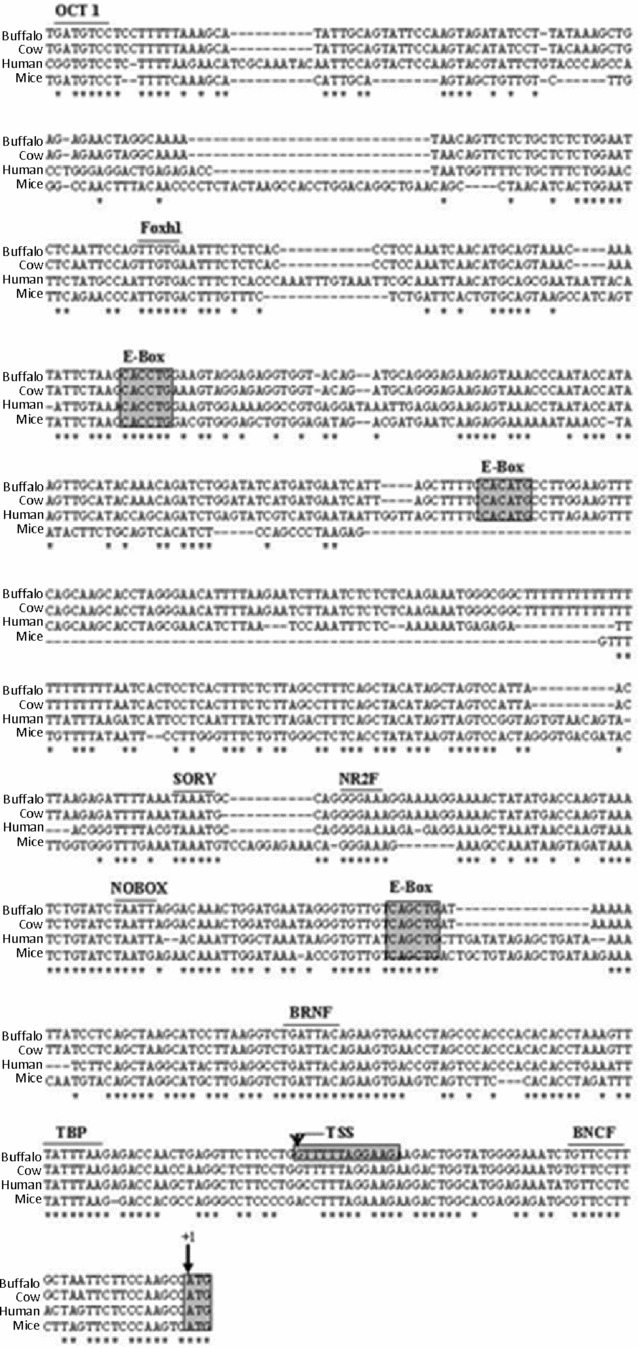
Figure 6 Homology analysis of GDF9 5′ upstream/promoter sequences of buffalo (FJ529502), cow (ENSBTAG00000009478), human (ENSG00000164404) and mouse (ENSMUSG00000018238). Asterisks represent identical nucleotides. Translational start site is indicated by +1. Putative germ cell-specific TF binding sites and cis-elements are labelled above the sequence.
Discussion
GDF9 gene expression has been reported in oocytes from a variety of rodent and mammalian species including murine, ovine, bovine, human and buffalo (McGrath et al., Reference McGrath, Esquela and Lee1995; Aaltonen et al., Reference Aaltonen, Laitinen, Vuojolainen, Jaatinen, Horelli-Kuitunen, Seppa, Louhio, Tuuri, Sjoberg, Butzow, Hovata, Dale and Ritvos1999; Bodensteiner et al., Reference Bodensteiner, Clay, Moeller and Sawyer1999; Hayashi et al., Reference Hayashi, Mcgee, Min, Klein, Rose, Duin and Hsueh1999; Sendai et al., Reference Sendai, Itoh, Yamashita and Hoshi2001) However, there is little information on the characteristics of this gene, or its control elements including the nature of regulatory control in buffalo.
In this study, we have characterized for the first time the oocyte-expressed complete GDF9 cDNA in in vitro matured oocytes from Indian river buffalo (Bubalus bubalis). The 5′ upstream sequence of GDF9 gene, its TSS, the core promoter element and putative cis-acting regulatory elements involved in its oocyte-specific expression were also described in this study. The deduced amino acid sequence of buffalo GDF9 revealed 10 hydrophobic amino acids at the NH2 terminus common for secretary proteins. Buffalo GDF9 amino acid sequence has 318 amino acids of proregion and 135 amino acids of mature protein that predict a molecular mass of 15.5 kDa for mature protein. Our data correspond well with earlier studies that reported that GDF9 is composed of pro- and mature regions separated by a tetrabasic cleavage sites (McPherron & Lee, Reference McPherron and Lee1993; Hayashi et al., Reference Hayashi, Mcgee, Min, Klein, Rose, Duin and Hsueh1999; Sendai et al., Reference Sendai, Itoh, Yamashita and Hoshi2001). Presence of a furin-like proteases site of bibasic sequence (R–H–R–R) that stretched from amino acids 315–318 on buffalo GDF9 further corresponded with the GDF9 protein structure reported in human (McPherron & Lee, Reference McPherron and Lee1993). Comparison of amino acid sequence of buffalo GDF9 cDNA with other vertebrates (Fig. 3) indicated that the amino acid sequence of mature protein is highly conserved among the species. The number and position of three asparagine residues (responsible for potential N-glycosylation) and eight cysteine residues are also conserved in the 453-amino-acid-containing peptide out of which one asparagine and six cysteine residues were located in the mature region (Fig. 2). These conserved cysteine residues present in the mature region have been reported to be responsible for the cysteine knot that results in the characteristic fold of the monomer, thereby making it biologically active. (Avsian-Kretchmer & Hsueh, Reference Avsian-Kretchmer and Hsueh2004). Differential level of GDF9 expression observed in different follicular size oocytes raises the possibility of the presence of transcript variants at the TSS level as observed in mouse ovarian tissue by Incerti et al. (Reference Incerti, Dong, Borsani and Matzuk1994), in which seven putative TSS were detected between 31 and 57 bp upstream of GDF9. In contrast, in buffalo, a single TSS was revealed located at 57 nucleotides upstream of the ATG. In RACE experiments, a heterogeneous population of in vitro matured oocytes collected from different follicular sizes was processed as a pool for RNA recovery. Sequence analysis of multiple clones that contained the RACE inserts confirmed the presence of single TSS in the buffalo GDF9 gene, irrespective of the follicular origin of oocytes. This situation signifies that differential expression of GDF9 in buffalo oocytes is not dependent on the variability of transcripts, but that the presence of some other potent regulatory elements in the 5′ upstream region may be crucial for up- and down-regulation of the oocyte-expressed GDF9 gene. The oocyte-expressed bubaline cDNA sequence reported in this study has been deposited in the NCBI GenBank database (accession no. FJ529501).
We isolated and sequenced the 4244-bp buffalo GDF9 5′ flanking region (NCBI GenBank accession no. FJ529502). As the consensus binding site of a transcription factor often consists of a very short stretch of DNA readily found in multiple sequences, interpretation based on in silico analysis might contain many false positives (Quandt et al., Reference Quandt, Frech, Karas, Wingender and Werner1995). Comparison of promoter sequences among different species offers one way of narrowing down such spurious conclusions. Therefore, we compared the 5′ upstream sequence of buffalo with other available sequences from cow, human and mouse to draw meaningful conclusions. As shown in Fig. 6, the 753-bp proximal region of the buffalo, cow, human and mouse GDF9 gene were found to be highly conserved. As mentioned before, a potential TATA element was detected at –23 nucleotides upstream of the mapped TSS, which signified that this stretch was qualified to be a possible core promoter element for the GDF9 gene.
Alignment of the proximal conserved region of the buffalo GDF9 gene revealed multiple conserved E-box elements. An E-box motif (CANNTG) has been detected in the promoter of many tissue-specific genes, namely the Zp genes and M6P/IGFIIR (Liang et al., Reference Liang, Soyal and Dean1997; Weiner et al., Reference Weiner, Chen and Davis2000). The consensus sequence CANNTG can bind to the proteins of a basic helix–loop–helix (bHLH) family to regulate transcription in a tissue-specific manner. Two nucleotides at the centre of CA and TG (CANNTG) of E-box motif have been reported for discriminatory binding among the different bHLH family members (Weintraub et al., Reference Weintraub, Genetta and Kadesch1994) which might lead to a variable expression pattern of its corresponding gene either in a temporal or in a spatial manner. With the same logic, we assumed the central NN combination as CA, CC and GC, which revealed at least three conserved E-box elements in buffalo, human and cow, although in mouse two out of these three E-boxes were present (Fig. 6). In the mouse, the E-box present within 200 bp of the ATG has been demonstrated to play a critical role in the expression of GDF9 in ovary (Yan et al., Reference Yan, Elvin, Lin, Hadsell, Wang, DeMayo and Matzuk2006). Thus we can speculate that in buffalo also the E-box mapped at position –170 to –165 bp can be validated for its major control on GDF9 expression. However, the relevance of other two conserved E-boxes found on buffalo sequence at the –520 to –525 and the –718 to –723 bp position also deserve attention in view of cases in which an E-box located at –1115 bp was found to be critical for muscle-specific enhancer function of sarcoplasmic reticulum (SR) Ca2+-ATPase gene (SERCA2) in rabbit; species specificity is also considered important in evolutionary origin of such controlling elements (Baker et al., Reference Baker, Dave, Reed, Misra and Periasamy1998).
In addition to E-boxes, we found two putative NBE (TAATTA) at –3471 and –203 positions from ATG. The NBE has been shown to be important for GDF9 expression in mice and has a sequence motif of TAATTG (Choi & Rajkovic, Reference Choi and Rajkovic2006). However Tsunemoto et al. (Reference Tsunemoto, Anzai, Matsuoka, Tokoro, Shin, Amano, Mitani, Kato, Hosoi and Saeki2008) reported that the NOBOX DNA binding elements had a role in oocyte-specific gene expression with consensus sequence 5′-TAATTG/A-3′; more than one NBE could have a synergistic role in control of the basal level of transcription for oocyte-specific genes. In that light, the 5′-TAATTA-3′ motifs found in buffalo sequence could be considered to be potential NBEs and its regulatory role in buffalo would be worth exploring.
In conclusion, this study is the first to report cloning and characterization of GDF9 full-length cDNA expressed in buffalo in vitro matured oocytes. In addition, we have characterized the 5′ upstream region of this gene. Complete cDNA sequence of the GDF9 gene with a single TSS, which is unique for buffalo, along with its full-length 3′UTR, was also worked out in this study. This study also reports a description of the 5′ upstream region of a nearly 5 kb fragment, which leads to identification of some potential oocyte-specific cis-acting elements such as E-boxes and NBEs. This study will provide important clues for optimization of the ART protocols in this important dairy animal.
Acknowledgements
This work was supported under the World Bank NAIP project C4056 and NAE projects of ICAR to the corresponding author.